
| Glioblastoma | |
|---|---|
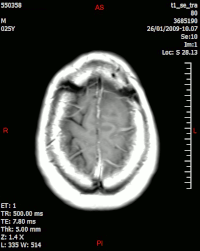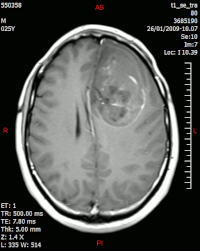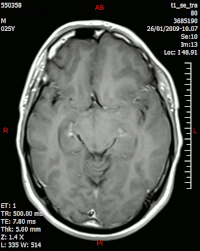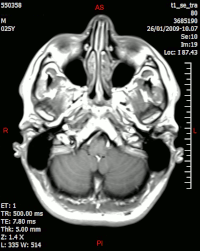
 Nell'immagine RM si noti l'anello di enhancement (la parte più evidente del tumore) intorno all'area centrale di necrosi. | |
| Tipo | Maligno |
| Fattori di rischio | radiazione ionizzante |
| Età media alla diagnosi | 53 anni |
| Rapporto M:F | 1,5:1 |
| Sigle | GBM |
| Sinonimi | |
| Glioblastoma multiforme, Astrocitoma di grado IV | |
| Classificazione e risorse esterne | |
| Neurooncologia | |
| Tumori del tessuto neuroepiteliale └► Tumori astrocitari └► Glioblastoma | |
| ICD-O | 9440/3 |
| Grado WHO | IV |
Il glioblastoma (noto anche come glioblastoma multiforme o con la sigla GBM, meno comunemente come glioblastoma polimorfo) è il tumore più comune e più maligno tra le neoplasie della glia. Il suo nome è stato stabilito dallo standard WHO-2000 e confermato dallo standard WHO-2007.
Composto da un eterogeneo insieme di cellule tumorali astrocitiche scarsamente differenziate, il glioblastoma colpisce soprattutto gli adulti, e si presenta solitamente negli emisferi cerebrali; meno frequentemente al tronco cerebrale o al midollo spinale. Come tutti i tumori cerebrali, salvo rarissimi casi, non si espande oltre le strutture del sistema nervoso centrale.
Il glioblastoma può svilupparsi da un astrocitoma diffuso (grado II) o da un astrocitoma anaplastico (grado III) (in tal caso è detto secondario, vedi più oltre), ma più frequentemente si manifesta de novo, senza alcuna evidenza di precedente neoplasia (è allora detto primario). Il trattamento del glioblastoma include chirurgia, radioterapia e chemioterapia. È difficile da curare e sono pochi i casi di sopravvivenza oltre i tre anni.
Classificazione
Stando alla penultima classificazione dei tumori del sistema nervoso centrale (2007) il glioblastoma fa parte dei tumori astrocitari, insieme con altri sei tipi di neoplasie: nella colonna a destra viene riportata la denominazione ufficiale del WHO-2007, insieme al codice ICD-O (International Classification of Diseases for Oncology: Classificazione Internazionale delle Malattie per l'Oncologia). Viene poi aggiunto il grading, come "WHO grade" seguito da numerazione romana. Della definizione italiana si riporta la versione più comunemente usata in letteratura. Altre, eventualmente presenti, sono in parentesi quadra. Le varianti sono indicate in corsivo.
| Astrocitoma pilocitico |
Pilocytic Astrocytoma, IDC-O 9421/1, WHO grade I
Pilomyxoid Astrocytoma, IDC-O 9425/3, WHO grade II |
| Astrocitoma subependimale a cellule giganti [Astrocitoma gigantocellulare subependimale] | Subependymal Giant Cell Astrocytoma, IDC-O 9384/1, WHO grade I |
| Xantoastrocitoma pleomorfo | Pleomorphic Xanthoastrocytoma, ICD-O 9424/3, WHO grade II |
| Astrocitoma diffuso [Astrocitoma] | Diffuse Astrocytoma, IDC-O 9400/3, WHO grade II |
| Astrocitoma anaplastico | Anaplastic Astrocytoma, IDC-O 9401/3, WHO grade III |
| Glioblastoma [Glioblastoma Multiforme, Astrocitoma di grado IV] |
Glioblastoma, IDC-O 9440/3, WHO grade IV
Giant Cell Glioblastoma, IDC-O 9441/3, WHO grade IV Gliosarcoma, IDC-O 9442/3, WHO grade IV |
| Gliomatosis cerebri | Gliomatosis Cerebri, IDC-O 9381/3, WHO grade III |
Storia

Nella prima metà del XIX secolo il glioblastoma era considerato di origine mesenchimale e quindi definito con il termine di sarcoma. Nel 1863 Rudolf Virchow ne stabilì l'origine gliale. F.B. Mallory, in una memoria del 1914, propone il termine glioblastoma multiforme. Si deve però attendere il 1925 per una descrizione compiuta della neoplasia, da parte di J.H. Globus e I. Strass. A quest'epoca la denominazione più comune del tumore è spongioblastoma multiforme. Nel 1926 una pubblicazione di P. Bailey e H. Cushing ripropone con successo la dizione di Mallory. La Classificazione WHO del 2000 dei Tumori del sistema nervoso fissa infine il nome a glioblastoma. (Per un resoconto storico più dettagliato si rimanda a K.J. Zülch e a D.S. Russell e L.J. Rubinstein).
Nello sviluppo del concetto che il glioblastoma talvolta emerge per progressione e malignizzazione di una lesione di grado più basso, un ruolo determinante hanno avuto gli studi di H.J. Scherer (1940) e J.W. Kernohan et al. (1949). Questo punto di vista ha ricevuto un forte sostegno dagli studi di genetica molecolare, i quali hanno mostrato che esiste una caratteristica accumulazione sequenziale di alterazioni geniche dagli astrocitomi diffusi di grado II al glioblastoma (Vedi più oltre, sezione Patogenesi, Tabb. 1 e 2).
Epidemiologia
Il glioblastoma è il tumore cerebrale più frequente, coprendo approssimativamente il 12-15% di tutte le neoplasie intracraniche e il 50-60% di tutti i tumori astrocitari (Vedi Classificazione).
Nella maggior parte dei paesi europei e del Nord America, l'incidenza è di 2-3 nuovi casi all'anno su 100 000 abitanti.
Il glioblastoma può manifestarsi a qualsiasi età, ma di preferenza si presenta negli adulti, con un picco tra i 45 e i 70 anni. Circa i due terzi dei pazienti (70%) ha un'età compresa nell'intervallo suddetto. L'età media è di circa 53 anni, con un rapporto Maschi/Femmine pari a 1,5:1. Questi ultimi dati provengono da un lavoro relativo a 1003 biopsie per glioblastoma, a cura dell'Ospedale Universitario di Zurigo. Sono citati in P. Kleihues et al. (2000). Dati simili sono riportati da altri autori.
In un lavoro relativo a 488 casi, G.J. Dohrman e altri hanno evidenziato che sono pediatrici l'8,8% dei glioblastomi. Rari sono invece i casi di glioblastomi congeniti, anche se diagnosi di glioma maligno tramite ecografia in utero hanno mostrato che il glioblastoma prenatale può manifestarsi anche a 29 settimane di gestazione.
I glioblastomi si presentano più spesso nella materia bianca subcorticale degli emisferi cerebrali. I siti più frequentemente affetti sono il lobo temporale (31%), il lobo parietale (24%), il frontale (23%) e l'occipitale (16%). La combinazione fronto-temporale è tipica. La neoplasia sovente si estende per infiltrazione alla corteccia adiacente, ai gangli della base e quindi all'emisfero controlaterale. Questi dati provengono da un rapporto relativo a 987 glioblastomi, a cura dell'Ospedale Universitario di Zurigo. Sono citati in P. Kleihues et al. (2000).
Glioblastomi intraventricolari sono eccezionali, mentre glioblastomi del tronco encefalico sono poco frequenti, anche se spesso riguardano i bambini. Il cervelletto e la colonna vertebrale raramente sono affetti da questa neoplasia.
Eziologia
Visione tradizionale
I tumori si formano in seguito a una crescita anormale e sregolata di cellule. Una volta che il cervello umano completa il suo sviluppo, subito dopo la nascita, la vasta maggioranza delle sue cellule entra in uno stato di quiescenza, nel quale non si dividono più. Unica eccezione a questa regola si ha quando si sviluppa un tumore.
Le cellule cerebrali tumorali riprendono il “ciclo cellulare” a causa di alterazioni in alcuni dei numerosi geni che controllano la divisione cellulare e la crescita. Benché molto sia noto sulle alterazioni di questi geni nei tumori cerebrali, la ragione prima per la quale insorgono le alterazioni attualmente è sconosciuta.
Ereditarietà
Si osservi che quando si parla di geni, non vuol dire che i tumori cerebrali siano ereditari. Benché ci siano delle sindromi nelle quali tali tumori presentano familiarità, queste situazioni (neurofibromatosi, sindrome di Turcot, sindrome di Li-Fraumeni, ecc.) sono molto rare e normalmente note in famiglia prima che si sviluppi un tumore in un membro familiare.
Fattori di rischio
La radiazione ionizzante è l'unico inequivocabile fattore di rischio che sia stato identificato per le neoplasie gliali e meningee. L'irradiazione del cranio, anche a basse dosi, può aumentare l'incidenza di tumori gliali di un fattore da 3 a 7 e di meningiomi di un fattore 10, con un periodo di latenza da 10 a più di 20 anni dopo l'esposizione.
Nessun'altra situazione ambientale o comportamento da parte del paziente è stata chiaramente identificata come fattore di rischio. Viene da più parti riportato che uso di telefoni cellulari, vicinanza a cavi di alta tensione, uso di coloranti per capelli, trauma cranico, alimentazione contenente N-nitrosammine, ovvero altri fattori nutrizionali, tutti incrementano il rischio di tumori cerebrali; tuttavia tali dati sono giudicati in conflitto e non convincenti.
L'associazione tra tipo di occupazione professionale e comparsa di glioblastomi è stata oggetto di numerosi studi. Lavoratori cronicamente esposti al cloruro di vinile, composti con base fenolica e idrocarburi aromatici sono risultati soggetti più a rischio.
Cellule staminali neoplastiche del cervello

A partire dagli anni novanta studi prima sugli animali e poi sull'uomo hanno mostrato che all'interno del cervello c'è una continua produzione di nuove cellule. In particolare nel giro dentato dell'ippocampo e nella zona subventricolare dei ventricoli laterali sono state individuate delle cellule staminali neuronali multipotenti, in grado cioè di produrre nuove cellule indifferenziate (staminali) e cellule mature, quali neuroni, astrociti e oligodendrociti. Sono anche capaci di auto-rinnovamento, in tal modo fanno sì che il numero totale delle cellule rimanga costante. D'altra parte, un filone della ricerca ha scoperto, a partire dal 2002, che nei tumori cerebrali, in particolare nei glioblastomi, esiste una gerarchia di cellule tumorali. Nel senso che una (piccola) parte del tumore è fatta da cellule che hanno le stesse caratteristiche delle staminali neuronali, talché gli autori hanno coniato il nome di cellule staminali neoplastiche del cervello (brain tumor stem cells). Sono queste il motore del tumore: riproducono in continuazione cellule staminali tumorali e cellule tumorali (non staminali). E sono solo le ultime a essere soggette agli attacchi delle terapie. Le staminali neoplastiche sono di fatto refrattarie a radioterapia e chemioterapia, in quanto capaci di auto-riparare in tempo i danni effettuati dalle terapie tradizionali, prima che i danni divengano irreversibili e tali da rendere inattiva la cellula. Basta quindi che una sola staminale cerebrale neoplastica sfugga alla chirurgia, perché si rimetta in moto il meccanismo e si abbia una ripresa di malattia. Si ipotizza che l'esistenza di queste cellule staminali neoplastiche del cervello emerga da un errore nella autoregolazione delle staminali neuronali (auto-rinnovamento), di cui si è detto prima. Lo schema concettuale qui riportato in forma molto succinta, in letteratura prende il nome di "Ipotesi delle cellule staminali neoplastiche". Tale schema è seguito dalla stragrande maggioranza dei ricercatori. Ciononostante esiste una piccola ma agguerrita minoranza che tende a dare una spiegazione differente dei fenomeni descritti o a inserirli in una cornice concettuale diversa.
Patogenesi
Nel seguito viene riportata schematicamente la sequenza di alterazioni genetiche che conducono al glioblastoma, come descritta nelle due ultime edizioni della classificazione WHO dei tumori del SNC.
Distinguiamo due tipi di alterazioni:
- l'attivazione di fattori oncogeni:
- EGF/R (Epidermal Growth Factor/Receptor, fattore di crescita dell'epidermide)
- MDM2 (L'oncoproteina Mouse Double Minute 2 promuove la sopravvivenza della cellula e la progressione del ciclo cellulare inibendo l'oncosoppressore TP53)
- PDGF/R (Platelet-Derived Growth Factor/Receptor, fattore di crescita derivato dalle piastrine)
- la disattivazione di fattori oncosoppressori:
- 10p, 10q, 19q (cromosomi)
- DCC (Deleted in Colorectal Cancer tumor suppressor gene, Gene con delezione nel cancro del colon-retto)
- p16 (Tumor suppressor gene/protein, Antigene tumorale cellulare)
- TP53 (Tumor suppressor gene/protein, Antigene tumorale cellulare)
- PTEN (Phosphatase and TENsin homolog è un oncosoppressore che controlla la crescita, la proliferazione e la sopravvivenza cellulare. Dalla sua mutazione o inibizione può conseguire l'insorgenza di tumori, ad es., della prostata, della mammella, del colon e del cervello.)
- RB (RetinoBlastoma tumor suppressor gene, proteina del retinoblastoma)
La Tabella 1 (P. Kleihues e H. Ohgaki, 1999,
come riportata in P. Kleihues et al., 2000, con modifiche grafiche) è tratta dalla classificazione WHO del 2000 e
mostra le mutazioni che avvengono da cellule sane al glioblastoma.
Nella parte a sinistra si può vedere l'attivazione delle lesioni intermedie (astrocitoma diffuso e astrocitoma anaplastico) prima di giungere al glioblastoma cosiddetto secondario (questo aggettivo qui non ha il significato solito di metastatico bensì di derivato da precedenti lesioni). Nella parte a destra la tabella mostra le mutazioni che dalle cellule sane portano direttamente (de novo) al glioblastoma, detto quindi primario.
(Tra parentesi è la percentuale di presenza della singola alterazione.)
| Tab. 1 Alterazioni genetiche del Glioblastoma (2000) | |
| Cellule astrocitiche differenziate ovvero precursori neuroepiteliali | |
|
Mutazione di TP53 (>65%) Sovraespressione di PDGF-A, PDGFR-α (~60%) ↓ |
EGFR: Amplificazione (~40%) Sovraespressione (~60%) MDM2: Amplificazione (<10%) Sovraespressione (~50%) Delezione di p16 (30-40%) Perdita di eterozigosi su 10p e 10q Mutazione di PTEN (~30%) Alterazione di RB ↓ |
| Astrocitoma diffuso | |
| Perdita di eterozigosi su 19q (~50%) Alterazione di RB (~25%) ↓ | |
| Astrocitoma anaplastico | |
| Perdita di eterozigosi su 10q Mutazione di PTEN (5%) Perdita di espressione di DCC (~50%) Amplificazione di PDGFR-α (<10%) ↓ | |
| Glioblastoma secondario | Glioblastoma de novo |
La Tabella 2 (da P. Kleihues et al., 2007, con modifiche grafiche) è tratta dalla Classificazione WHO del 2007. Sono riassunti ulteriori anni di studi e di approfondimento.
Si osservi che l'asterisco (*) segnala alterazioni genetiche che sono significativamente differenti in frequenza tra glioblastomi primari e secondari.
| Tab. 2 Alterazioni genetiche del glioblastoma (2007) | ||
| Cellule astrocitiche differenziate ovvero precursori ovvero staminali | ||
| Mutazione di TP53 (59%) ↓ |
Mutazione di TP53 (59%) ↓ |
I I I I ↓ <3 mesi (68%) <6 mesi (84%) I I I I ↓ |
| Astrocitoma diffuso | Astrocitoma diffuso | |
| Mutazione di TP53 (53%) ↓ |
I I ↓ 5,1 anni I I ↓ |
|
| Astrocitoma anaplastico | ||
| ↓ 1,9 anni ↓ | ||
| Perdita di eterozigosi su 10q (63%) Amplificazione di EGFR (8%) Delezione di (19%) Mutazione di TP53 (65%)* Mutazione di PTEN (4%) ↓ |
Perdita di eterozigosi su 10q (70%) Amplificazione di EGFR (36%)* Delezione di (31%) Mutazione di TP53 (28%) Mutazione di PTEN (25%)* ↓ |
|
| Glioblastoma secondario | Glioblastoma primario | |
| 5% dei casi Età media: 45 anni Rapporto M/F: 0,65 |
95% dei casi Età media: 62 anni Rapporto M/F: 1,33 |
|

I collegamenti tra le due tabelle si possono desumere esaminando i riferimenti bibliografici indicati.
Un fatto però è necessario evidenziare. Un esame, anche superficiale di queste tabelle, porta alla conclusione che i glioblastomi primari e secondari sono due malattie distinte (anche se istologicamente poco distinguibili), che colpiscono gruppi di pazienti diversi per età e sesso e che si sviluppano attraverso cammini genetici diversi, con diversi profili di espressione proteica e mRNA. Queste differenze sono importanti, specialmente in quanto possono influenzare la risposta del tumore alla radio- e chemio-terapia e possono costituire il target di futuri approcci terapeutici.
Complicanze
Si riporta nel seguito un elenco sommario delle complicanze legate al glioblastoma, distinguendo quelle dovute alla malattia da quelle più legate alle terapie.
Molte di tali complicanze non sono comuni e un numero significativo di esse può essere tenuto sotto controllo terapeuticamente in modo efficace.
- Complicanze legate al tumore:
- Edema cerebrale
- Disturbi neurologici
- Disturbi visivi
- Idrocefalo
- Gliomatosi leptomeningea
- Deterioramento delle funzioni cognitive
- Deterioramento dello stato psicologico (angoscia, ecc.)
- Complicanze legate alle terapie:
- Patologie legate alla chirurgia
- Infezioni
- Disturbi neurologici
- Disturbi visivi
- Patologie legate alla radioterapia
- Disturbi neurologici
- Disturbi visivi
- Deterioramento delle funzioni cognitive
- Patologie legate alla chemioterapia
- Disfunzioni ematiche
- Disturbi dell'apparato respiratorio
- Diarrea
- Spossatezza
- Disturbi neurologici
- Disfunzioni ematiche
- Patologie legate ai farmaci anticonvulsivanti
- Patologie legate ai farmaci antinfiammatori
- Patologie legate ai farmaci citostatici
- Patologie legate alla chirurgia
Anatomia patologica
Esame macroscopico

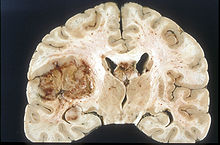
Nonostante la breve durata dei sintomi, i glioblastomi sono spesso di grandi dimensioni al momento della presentazione e possono occupare più di un lobo.
La lesione è in genere monolaterale, ma quelle del tronco cerebrale e del corpo calloso possono essere bilateralmente simmetriche. Il tumore occupa la stessa posizione nei due emisferi e si presenta con un aspetto "a farfalla".
L'estensione sopratentoriale bilaterale è dovuta a una rapida crescita lungo le strutture mielinizzate, in particolare attraverso il corpo calloso e lungo i fornici verso i lobi temporali.
I confini della massa neoplastica, che non è mai capsulata, sono ovunque sfumati.
Il colorito è grigiastro, ma si possono reperire abbondanti variegazioni, provocate da necrosi
o da emorragie più o meno recenti, per cui sullo sfondo grigio compaiono aree giallastre, per degenerazione grassa o per necrosi e aree rossastre o nerastre dovute alle emorragie.
La zona periferica di tessuto tumorale ipercellulare appare come una rima morbida e grigia.
Il tessuto necrotico può anche bordare strutture cerebrali adiacenti senza una zona intermedia tumorale macroscopicamente rilevabile.
La necrosi centrale può occupare più dell'80% della massa totale del tumore.
I glioblastomi sono tipicamente punteggiati di macchie di color rosso e marrone per le emorragie, le quali a volte sono abbastanza ampie da provocare sintomi simili al colpo apoplettico, che può costituire il primo segno clinico del tumore.
Cisti macroscopiche, quando presenti, contengono un fluido torbido proveniente da tessuto tumorale necrotico liquefatto, nettamente in contrasto con le ben delineate cisti di ritenzione degli astrocitomi diffusi di grado II.
La maggior parte dei glioblastomi degli emisferi cerebrali è chiaramente intraparenchimale, con epicentro nella sostanza bianca. Qualche volta la neoplasia si presenta come ampiamente superficiale e in contatto con leptomeningi e dura madre e può essere scambiata per un carcinoma metastatico ovvero per una lesione extra-assiale come il meningioma.
Esame microscopico


Il glioblastoma è una neoplasia anaplastica della glia costituita da cellule tumorali astrocitiche scarsamente differenziate, polimorfe, con marcate atipie nucleari e una intensa attività mitotica. Caratteristiche peculiari ai fini diagnostici sono pure la cospicua proliferazione microvascolare e la presenza di necrosi. Come suggerisce l'aggettivo “multiforme” del sinonimo più comune, la morfologia istologica del glioblastoma è estremamente variabile, con cellule rotondeggianti, a forma di fuso, di dimensioni piuttosto piccole o molto grandi.
Mentre alcuni glioblastomi mostrano un alto grado di polimorfismo cellulare e nucleare, con numerose cellule giganti plurinucleate, altri presentano una conformazione caratterizzata da intensa cellularità, ma piuttosto ripetitiva. La natura astrocitica della neoplasia può risultare abbastanza facile da identificare, almeno localmente, in alcuni tumori, ma difficile da riconoscere in altri, a causa dell'alto grado di anaplasia. L'eterogeneità da regione a regione del glioblastoma è rilevante e rende un'impresa difficile effettuarne la diagnosi su campioni limitati quali quelli ottenuti tramite biopsia stereotassica (Si veda l'illustrazione nella sezione Chirurgia.). Quantunque prevalga la presenza di cellule scarsamente differenziate, in alcuni punti possono essere distinguibili astrociti neoplastici più differenziati. Questo è particolarmente vero nei casi di glioblastoma derivante da progressione di un astrocitoma diffuso (Grado II della scala WHO). La transizione tra aree che ancora posseggono differenziazione astrocitica riconoscibile e aree ad alta anaplasia cellulare può essere continua o repentina. Una brusca variazione nella morfologia riflette di solito la comparsa di un tumore diverso, nato per acquisizione di una o più alterazioni genetiche aggiuntive.
Nel contesto della neoplasia si osservano vaste aree di necrosi, circondate da nuclei disposti parallelamente tra loro, costituendo tipiche “palizzate”. Si riscontra una marcata proliferazione di cellule endoteliali con formazione di numerosi vasi, alle volte con aspetto simile ad ammasso o gomitolo. Alcuni hanno parete ialina e altri sono trombizzati. La proliferazione endoteliale comunque non è diffusa, ma è focalizzata in alcuni punti. Attorno alla neoplasia si possono rinvenire aree di astrociti gemistocitici (astrocitomi diffusi di grado II).
Clinica
Segni e sintomi
La storia clinica della malattia normalmente è breve (meno di 3 mesi, in più del 50% dei casi), a meno che il tumore non si sviluppi per progressione da un astrocitoma di basso grado (glioblastoma secondario; vedi più sopra la sezione Patogenesi).
I sintomi del glioblastoma sono quelli aspecifici di una massa in espansione all'interno del cranio, quindi di una crescente pressione endocranica. Comuni sono cefalea, nausea, vomito, dilatazione dei vasi cerebrali con alterazioni della retina fino al papilledema, emiparesi, emianestesia, emianopsia, diplopia, afasia e crisi convulsive.
La percentuale di pazienti soggetti ad attacchi epilettici arriva sino a un terzo. Da segnalare infine sintomi neurologici non specifici quali l'obnubilamento della coscienza e modifiche della personalità.
Diagnostica per immagini e tumori cerebrali


La presenza di una neoplasia cerebrale può essere efficacemente rivelata attraverso la tomografia computerizzata (TAC) e la risonanza magnetica nucleare (RM).
La RM presenta una maggiore sensibilità rispetto alla TAC nella identificazione delle lesioni; tuttavia non è sempre di facile accesso per il paziente e presenta alcune controindicazioni: non può essere effettuata in portatori di pacemaker, di protesi incompatibili con il campo magnetico, di clips metalliche ecc. La TAC resta la metodica di elezione nella rivelazione di calcificazioni interne alle lesioni o di erosioni ossee della teca o della base cranica.
L'uso del mezzo di contrasto (iodato in caso di TAC, paramagnetico in caso di RM (gadolinio)), permette l'acquisizione di informazioni sulla vascolarizzazione e sull'integrità della barriera emato-encefalica, una migliore definizione del nodulo tumorale rispetto all'edema cerebrale circostante e consente di avanzare ipotesi sul grado di malignità.
L'esame radiologico permette inoltre di valutare gli effetti meccanici (e le conseguenti modificazioni dei rapporti delle strutture encefaliche) derivanti dalla presenza della massa “estranea”: idrocefalo ed ernie, i cui effetti possono anche risultare letali.
L'esame, infine, in vista dell'intervento chirurgico, precisa la sede della lesione e la vicinanza (o addirittura il coinvolgimento) del tumore in zone del cervello assolutamente vitali (aree cosiddette “eloquenti”). A questo scopo la RM risulta superiore alla TAC per il fatto che è in grado di fornire immagini tridimensionali.
Prima di chiudere questa sezione è utile richiamare l'attenzione su alcuni concetti e termini che risulteranno utili per la comprensione di sezioni successive.
Aspetto radiologico del tessuto neoplastico
Si vuole mettere in evidenza il fenomeno di alterazione dal punto di vista radiologico del tessuto neoplastico rispetto al normale parenchima cerebrale (modificazioni della densità elettronica dei materiali in caso di TAC e dell'intensità di segnale per la RM).
Come la maggior parte dei tessuti patologici, anche i tumori sono caratterizzati da un maggiore accumulo di acqua intracellulare. Alla TAC appaiono ipodensi, ovvero di densità inferiore al parenchima cerebrale, alla RM appaiono ipointensi nelle immagini T1-pesate e iperintensi in quelle DP- e T2-pesate. (Cfr. le voci Tomografia computerizzata e Imaging a risonanza magnetica).
Contrast enhancement (aumento di segnale di contrasto)
In una lastra radiografica la zona di cervello sano non dovrebbe segnalare particolari luminescenze. È naturale quindi che si ponga attenzione alle porzioni di maggior segnale di contrasto.
Nel tumore, in genere, la maggior quota di “contrast enhancement” è dovuta alla particolare barriera emato-tumorale, che permette il passaggio di iodio (TAC) e di gadolinio (RM) nello spazio interstiziale extravascolare intratumorale: aumenta così il segnale (densità o intensità) del tumore.
Si faccia comunque attenzione al fatto che il “contrast enhancement” non delimita con certezza il tumore dall'edema perilesionale: in effetti nei gliomi infiltranti maligni (quali, ad es., il glioblastoma e l'astrocitoma anaplastico) il reperto anatomo-patologico mostra tessuto neoplastico persino oltre l'edema vasogenico (quello cioè causato dalla distruzione della barriera emato-encefalica da parte del tumore), cosa questa che non è facilmente dimostrabile attraverso le immagini radiografiche.


Controllo post-chirurgico
Il controllo post-chirurgico tramite RM (o TAC) ai fini della determinazione della radicalità della rimozione di un tumore è ritenuto opinabile nella letteratura: l'esame dovrebbe essere eseguito entro 24 ore dalla chirurgia, prima cioè che si instaurino le alterazioni della barriera emato-encefalica sostenute dai fenomeni fibrotici-cicatriziali; in altre parole, la cicatrice fisiologica ha un “contrast enhancement” che può facilmente confondersi con un residuo o con una ricrescita di tumore.
Anche dopo trattamento radio-chirurgico, una radionecrosi (vedi più sotto la sezione omonima) può presentare caratteristiche di “imaging” e di “contrast enhancement” con aspetto quasi sovrapponibile a quello di un glioma maligno. Solo mediante metodiche funzionali come la tomografia a emissione di positroni (PET) con fluorodesossiglucosio (FDG-PET), la quale dimostra una più elevata captazione di glucosio da parte del tumore rispetto al tessuto sano, è possibile valutare l'assenza di metabolismo nelle necrosi rispetto alla recidiva tumorale, (anche se è possibile che necrosi e recidiva coesistano).
In alternativa alla PET si può utilizzare l'analisi spettroscopica mediante RM (Vedi Spettroscopia di risonanza magnetica nucleare e Risonanza magnetica funzionale): nella “mappa” dei metabolici di tale metodica è presente il picco della colina (Cho) che è associato alla sintesi delle membrane cellulari: un alto picco è indicativo di elevato turnover cellulare, come si verifica nei tumori.
Diagnostica per immagini e glioblastoma



La TAC evidenzia una lesione di morfologia irregolare, prevalentemente ipodensa, fortemente disomogenea per la presenza di vaste aree necrotiche di più netta ipodensità e di aree solide iperdense. Queste ultime sono espressione di un rapido accrescimento e conseguentemente di una elevata malignità.
Frequenti le zone emorragiche, variabili da piccoli foci a vaste aree ematiche che possono coinvolgere l'intera lesione.
Caratteristica è la morfologia a "farfalla" se presente l'interessamento di entrambi gli emisferi attraverso il corpo calloso.
Dopo contrasto compaiono grossolani cercini di impregnazione intorno alle aree necrotiche.
Alla RM la parte solida appare ipointensa in T1 e iperintensa in T2 con zone di segnale più elevato nelle parti a più forte cellularità. Le aree necrotiche, sempre iperintense in T2, possono presentarsi ipo-, iso- o iperintense in T1 in funzione del contenuto proteico o di prodotti di degradazione dell'emoglobina.
L'enhancement dopo mezzo di contrasto è in genere intenso e irregolare alla periferia del tumore e identifica soprattutto la componente cellulare “proliferativa” della neoplasia.
Comuni le aree puntiformi e serpiginose di assenza di segnale da flusso connesse alla presenza della ricca neovascolarizzazione. Questi vasi patologici neoformati sono privi di barriera ematoencefalica: ciò spiega sia l'impregnazione abbondante e grossolana sia l'edema vasogenico perilesionale (vedi la sezione precedente) dovuto al passaggio di liquido in sede extracellulare.
Diagnosi differenziale
La diagnosi differenziale si pone con: metastasi, emorragie cerebrali spontanee, ascessi, forme atipiche di sclerosi multipla, danno di barriera secondario a radioterapia.
Diagnostica per immagini. Conclusioni
Si può concludere arguendo che il primo passo da considerare nella valutazione di un paziente nel quale si sospetti una neoplasia cerebrale è la risonanza magnetica.
Tale esame sarebbe altresì da consigliare a ogni paziente che soffra di crisi epilettiche, per le quali non vi siano immediate e plausibili giustificazioni.
Normalmente la risonanza rivela senza particolari difficoltà la presenza del glioblastoma quale causa dei sintomi lamentati e non sono necessari ulteriori esami.
Trattamento
Nel trattamento del glioblastoma, come per qualunque altro tumore cerebrale, distinguiamo le terapie di supporto dalle terapie curative.
Terapie di supporto
Il trattamento di supporto ha come scopo di alleviare i sintomi e di migliorare le funzioni neurologiche del paziente. Gli agenti di supporto primari sono i farmaci antiepilettici e i corticosteroidi.
Farmaci antiepilettici

I farmaci antiepilettici vengono somministrati a circa il 25% dei pazienti che hanno avuto crisi epilettiche alla presentazione della malattia. La fenitoina (300–400 mg/d) è il farmaco più comunemente usato, ma carbamazepina (600-1 000 mg/d), fenobarbital (90–150 mg/d) e acido valproico (750-1 500 mg/d) sono ugualmente efficaci. Le dosi di tutti questi anticonvulsionanti devono essere adattate ai livelli che si riscontrano poi nel sangue del paziente, per fornire la massima protezione.
Similmente efficaci sono gli antiepilettici di nuova concezione, quali levetiracetam, gabapentin, lamotrigina e topiramato. La maggior parte di questi nuovi principi attivi ha il vantaggio di causare scarsi effetti collaterali di tipo cognitivo e, per il fatto che non inducono il sistema epatico microsomiale, non alterano il metabolismo dei chemioterapici. Questi nuovi anticonvulsivi stanno rapidamente sostituendo i farmaci classici nella terapia antiepilettica di prima linea.
Profilassi
Le sperimentazioni cliniche di tipo prospettico hanno dato risultati negativi nel tentativo di mostrare l'efficacia di un uso profilattico dei farmaci antiepilettici in caso di pazienti di tumori cerebrali che non avevano mai lamentato crisi epilettiche.
Di conseguenza, la letteratura medica ne sconsiglia l'uso per questo scopo eccetto che per il periodo relativo all'intervento chirurgico, quando il loro utilizzo può ridurre l'incidenza di crisi epilettiche post-operatorie.
Nei casi quindi di pazienti che non hanno mai avuto crisi è consigliabile che gli anticonvulsivi non siano più somministrati entro le 2 settimane dall'intervento.
Corticosteroidi
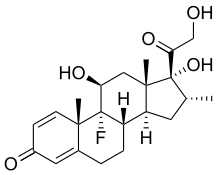
I farmaci a base di corticosteroidi sono in grado di ridurre l'edema peritumorale, diminuendo l'effetto massa della neoplasia e riducendo la pressione endocranica. Come effetto immediato si ottiene un sollievo al mal di testa e un miglioramento dei segni “lateralizzanti” (vedi la sintomatologia descritta alla voce epilessia).
Il corticosteroide di elezione è il desametasone, a motivo della sua minima attività mineralcorticoide.
La dose di partenza è di circa 16 mg/d. Tale quantità può essere aumentata o diminuita fino a raggiungere la minima dose necessaria per tenere sotto controllo la sintomatologia neurologica.
L'uso prolungato di corticosteroidi è associato a ipertensione, diabete mellito, stato iperglicemico iperosmolare non chetosico (affezione a pericolo di vita), miopatia, aumento di peso, insonnia e osteoporosi. Talché nel paziente di tumore cerebrale la dose steroidea dev'essere ridotta gradualmente “il più rapidamente possibile”, una volta che sia iniziato il trattamento curativo.
Per la maggior parte dei pazienti si cessa la somministrazione dei corticosteroidi quando hanno completato la radioterapia.
Ai pazienti sotto steroidi per più di 6 settimane si consiglia una profilassi antibiotica per la polmonite da pneumocystis carinii, cura che dovrebbe continuare per 1 mese dalla cessazione della somministrazione di corticosteroidi.
Terapie curative
Le terapie curative dei tumori cerebrali includono essenzialmente chirurgia, radioterapia e chemioterapia.
Il primo passo è, se possibile, di stendere un piano terapeutico generale che permetta di abbozzare la sequenza e i singoli elementi del trattamento multidisciplinare.
Chirurgia

L'approccio chirurgico dev'essere scelto accuratamente, allo scopo di ottenere la massima asportazione possibile del tumore, preservando le strutture vitali del cervello e minimizzando il rischio di deficit neurologico postoperatorio.
Gli obiettivi dell'intervento chirurgico sono:
- ottenere un'accurata diagnosi istologica;
- ridurre l'effetto massa sul cervello causato dal tumore e/o dall'edema peritumorale;
- se del caso, mantenere o ristabilire il flusso del liquido cefalorachidiano;
- conseguire una (potenziale) guarigione attraverso la rimozione “totale” della neoplasia (nel caso di glioblastoma, l'operazione chirurgica molto raramente ottiene la guarigione, comunque riduce le dimensioni del tumore in maniera tale da renderlo più gestibile da radio- e chemio-terapia).
Un'asportazione maggiore del 98% del volume del tumore (resezione “totale”) aumenta la sopravvivenza rispetto a una resezione subtotale o parziale. La resezione subtotale “estesa” non sembra conferire alcun vantaggio di sopravvivenza rispetto alla biopsia o alla resezione parziale.
In caso di ripresentazione della malattia (e questo avviene nella quasi totalità dei glioblastomi), o di espansione della parte di tumore rimasta dall'operazione chirurgica, o di radionecrosi (sia la ripresa di malattia sia la radionecrosi causano effetto massa ed edema e, come detto in precedenti sezioni, non sono distinguibili alla risonanza classica) si ricorre a un secondo intervento, per ridurre gli effetti della massa neoformata sul parenchima cerebrale.
In situazione di ricorrenza difficilmente si ottiene la guarigione, comunque ne consegue di solito un miglioramento della qualità della vita e una modesta estensione di sopravvivenza media.
In generale, un secondo intervento viene escluso in pazienti con indice di Karnofsky (KPS) inferiore o uguale a 60 o in quei pazienti che non sono candidabili a terapie adiuvanti successive alla chirurgia.
Prima di concludere questa sezione val la pena di accennare a studi clinici che prevedono durante l'operazione chirurgica la somministrazione intratecale di chemioterapici o immunoterapici ovvero di liquidi radianti. Questi studi sono nella fase di prima sperimentazione.
Il posizionamento sul letto operatorio di “wafers” impregnati di carmustina è l'unico caso di chemioterapia intracavitaria attualmente (settembre 2008) approvata dalla FDA (Food and Drug Administration) per il caso di glioblastoma.
Negli anni 2010, l'intervento chirurgico si è evoluto andando a rimuovere il tumore del pazienze in awake, ovvero "da sveglio". Grazie a tale operazione si può sondare se una determinata parte del cervello si può rimuovere senza che il paziente ne possa subire conseguenze dopo la sua operazione. Si elettro-stimolano specifiche zone del cervello per comprendere se il paziente risponde man mano a una psicologa la quale comunica con i chirurghi per capire fino a quando è possibile rimuovere il tumore.
Radioterapia
La radioterapia, che normalmente viene effettuata dopo l'operazione chirurgica, riguarda la parte di encefalo interessata dall'intervento oltre a un leggero margine esterno, e ha lo scopo di danneggiare il DNA di eventuali cellule tumorali rimaste dopo l'operazione e sfuggite al chirurgo perché non visibili al microscopio (in quanto infiltratesi più o meno distanti dalla zona dell'operazione).
Se la radioterapia riesce a danneggiare tali cellule prima che esse abbiano la possibilità di riparare il DNA e di riprendere la moltiplicazione cellulare, il paziente ne guadagna in sopravvivenza.
Sperimentazioni cliniche sui gliomi di alto grado (astrocitoma anaplastico, oligodentroglioma anaplastico, oligostrocitoma anaplastico, glioblastoma) effettuate dal BTSG (Brain Tumor Study Group) statunitense hanno mostrato che la radioterapia postoperatoria a dosi superiori a 50 Gy fornisce un miglioramento della sopravvivenza rispetto a nessun trattamento postoperatorio e che 60 Gy danno come risultato una sopravvivenza significativamente più lunga rispetto ai 50 Gy.
Questa quantità di radiazione corrisponde a una dose appena al di sopra di quella necessaria alla formazione di radionecrosi, perciò si è scelto come standard il trattamento radioterapico di 60 Gy complessivi somministrati in 30-33 frazioni, una al giorno.
Pazienti di glioblastoma di oltre 60 anni con una terapia abbreviata di 40 Gy in 15 frazioni mostrano sopravvivenza identica a quella ottenuta col regime standard. Perciò è ragionevole per tali pazienti l'uso di tale trattamento ridotto.
Circa la metà dei pazienti con astrocitoma anaplastico “risponde” alla radioterapia con 60 Gy (dato verificabile tramite evidenza radiografica); la percentuale si riduce al 25% per i pazienti con glioblastoma. Per entrambe le neoplasie casi di guarigione completa per radioterapia sono molto rari.
Nel tentativo di migliorare i risultati sunnominati, sono stati messi a punto un certo numero di nuovi approcci, quali la radioterapia iperfrazionata (HFRT), la brachiterapia (utilizzo di aghi radioattivi depositati direttamente), la radiochirurgia. Quest'ultima ha goduto nel recente passato di un certo interesse, in quanto trattasi di procedura non invasiva, da poter essere effettuata in certi casi anche in situazione di day-hospital. Richiede una selezione molto accurata dei pazienti, in quanto, tra l'altro, necessita che la neoplasia non sia estesa bensì altamente focalizzata.
A parte casi particolari, queste nuove tecniche non hanno mostrato di migliorare significativamente la sopravvivenza globale del paziente.
Radionecrosi
Si è già accennato in precedenti sezioni alla necrosi radioindotta. Questa complicanza è prodotta soprattutto dalla brachiterapia e dalla radiochirurgia e determina la sintomatologia da effetto massa, più sopra descritta, in circa il 50% dei pazienti di glioma maligno. Con il trattamento con corticosteroidi si riesce spesso a controllare l'edema circostante l'area radio-necrotica. Ciò, a lungo andare, a sua volta produce però dipendenza dagli steroidi, con tutte le complicazioni dell'uso prolungato a cui si è fatto cenno (nella sezione dei Corticosteroidi). Nei casi più gravi occorre far ricorso alla operazione chirurgica per rimuovere la massa necrotica.
Chemioterapia
Anche la chemioterapia ha lo scopo di danneggiare l'organizzazione del DNA delle cellule tumorali, eventualmente rimaste dopo l'operazione chirurgica e sfuggite alla radioterapia. Se il chemioterapico riesce a scardinare tale DNA, la cellula tumorale passa in fase di "morte programmata" (apoptosi).

La chemioterapia apporta benefici limitati ai pazienti di glioblastoma. Nelle sperimentazioni cliniche l'uso di nitrosuree non ha allungato significativamente la sopravvivenza media in tutti i pazienti, ma un sottogruppo di essi pare beneficiare di una sopravvivenza prolungata con l'aggiunta di chemioterapia alla radioterapia. Fattori prognostici quali l'età, l'indice di Karnofsy, ecc. non riescono a predire quali pazienti trarranno vantaggi dalla chemioterapia. (Vedi però, più oltre, il caso della temozolomide).
In una larga sperimentazione di fase III, i pazienti (con diagnosi di glioblastoma e senza alcun trattamento radio- chemio-terapico precedente) sono stati “randomizzati” per ricevere sola radioterapia (gruppo A) oppure radioterapia con contemporanea somministrazione giornaliera del farmaco temozolomide, seguita da somministrazione mensile sempre di temozolomide (gruppo B).
Sul totale di 573 pazienti la sopravvivenza media è passata da 12,1 mesi (gruppo A) a 14,6 mesi (gruppo B). Ma, cosa ancor più significativa, la sopravvivenza a due anni è più che raddoppiata, passando da 10,4% del gruppo A a 26,5% per il gruppo B.
Il trattamento in combinata radioterapia-temozolomide è risultato mediamente ben tollerato e con una tossicità aggiuntiva minimale, talché questo protocollo è diventato lo standard terapeutico di elezione per di tutti i nuovi pazienti di glioblastoma.
Come prodotto collaterale dello studio di cui si diceva, è stata individuata una proteina tumorale (MGMT) in grado di predire, con un'approssimazione utile nella pratica, quali pazienti beneficeranno del protocollo combinato. Questa metodica è ancora in fase di prova da parte della comunità scientifica e qui viene solo citata per informazione.
Cannabinoidi
Un discorso a parte meritano i cannabinoidi. Dei derivati della cannabis è nota l'efficacia in oncologia (attraverso capsule di tetraidrocannabinolo (THC) ovvero l'analogo sintetico nabilone), da un lato per combattere la nausea e il vomito indotti dalla chemioterapia, dall'altro per stimolare l'appetito e attenuare il senso di angoscia ovvero il dolore vero e proprio.
Dimostrata è la loro capacità di inibire la crescita e l'angiogenesi nei gliomi maligni.
I risultati di uno studio pilota relativo all'uso di THC su pazienti (in fase terminale) affetti da glioblastoma ricorrente sono apparsi meritevoli di approfondimento.
Ma estremamente interessante è la scoperta (per ora confermata su animali) che i cannabinoidi sono in grado di attaccare le cellule staminali neoplastiche del glioblastoma, col risultato da un lato di indurre la loro differenziazione in cellule più mature (e quindi più “trattabili”) e dall'altro di inibire la tumorigenesi.
Ricorrenza
Nonostante i (limitati) successi iniziali delle terapie, praticamente tutti i glioblastomi si ripresentano.
In tale situazione il paziente può essere sottoposto a una seconda operazione (se è nelle condizioni previste) ovvero può beneficiare di tecniche radioterapiche focalizzate (radiochirurgia, se la neoplasia risponde ai requisiti visti prima. Si osservi che, di solito, non è possibile effettuare un secondo ciclo di radioterapia standard a 60 Gy.), oppure gli possono essere somministrati chemioterapici diversi dalla temozolomide (a cui il paziente ”non risponde più”).

Tipici chemioterapici di ricorrenza sono la procarbazina, le nitrosuree, il melphalan, il carboplatino e altri.
In studi clinici recenti hanno mostrato un'attività antitumorale di interessante significatività l'utilizzo di mitoxantrone e il combinato di idrossiurea con imatinib mesilato.
Altre sperimentazioni cliniche suggeriscono l'impiego di inibitori dei recettori del fattore di crescita dell'epidermide o l'utilizzo di agenti anti-angiogenesi, ovvero di terapie combinate di radiofarmaci iniettati localmente insieme a chemioterapici pure iniettati localmente.
Tutti questi studi e protocolli sono al vaglio della comunità scientifica.
Un obiettivo comunque inseguito è l'individuazione di una metodica pratica per caratterizzare le classi di pazienti per le quali un protocollo dia i risultati migliori, in modo che assegnando il particolare paziente alla classe più opportuna gli venga praticato il protocollo di maggior efficacia, utilità e minimo impatto.
Prognosi
Sperimentazioni cliniche “randomizzate” del 1978 mostravano che la sopravvivenza media dopo la chirurgia, per pazienti in cura con soli corticosteroidi, era di 14 settimane, che salivano a 38 dopo la radioterapia. La chemioterapia allunga la sopravvivenza. Pazienti trattati con chirurgia, radioterapia e chemioterapia avevano una sopravvivenza media di circa 1 anno, salita a 15-18 mesi nel 2015.. Uno studio del 1998 riguardante 279 pazienti, che avevano ricevuto un trattamento completo aggressivo, riporta che solo 5 di essi (l'1,8%) sono sopravvissuti oltre i 3 anni.
In realtà ogni paziente reagisce in modo diverso alle terapie, talché per il singolo le probabilità di sopravvivenza (nel caso di trattamento completo, comprensivo di gestione della ricorrenza) risultavano nel 2008 pari al 57% a un anno, 16% a due anni e 7% a tre anni.. Dati del 2014 confermano una scarsa sopravvivenza oltre i 2,5 anni, con solo il 5% dei pazienti trattati che sopravvive a 5 anni dalla diagnosi, mentre per i pazienti non trattati la sopravvivenza media è di tre mesi dalla diagnosi. Dopo i 3 anni in letteratura si parla di "lunga sopravvivenza".
La sopravvivenza dopo 3 anni è rara, più frequente nel glioblastoma secondario. Un caso limite di lunga sopravvivenza (11 anni) è costituito dal famoso medico psichiatra francese David Servan-Schreiber, sopravvissuto prima 8 anni ad astrocitoma di IV stadio e poi 11 anni a glioblastoma secondario di I stadio da esso derivato, per un totale di 19 anni di vita in buona qualità, dopo la diagnosi di tumore che nel 1992 gli aveva lasciato pochi mesi. Grazie a cure sperimentali, sopravvisse 1 anno al glioblastoma di IV stadio ai lobi frontali, in discrete condizioni (2010-2011).
Un dato importante deriva da uno studio del 2003: la probabilità di sopravvivere per un altro anno, essendo già sopravvissuti uno, due, tre, quattro o cinque anni dopo la craniotomia è, rispettivamente, del 64,8%, 58,7%, 85,7%, 80,0%, 75,0%.








Voci correlate
- Classificazione dei tumori del sistema nervoso centrale
- Gradazione dei tumori del sistema nervoso centrale
- Neurooncologia
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su glioblastoma
Wikimedia Commons contiene immagini o altri file su glioblastoma
Collegamenti esterni
|
Classificazione e risorse esterne (EN) |
ICD-10: C71; OMIM: 137800; MeSH: D005909; DiseasesDB: 29448; |
| Controllo di autorità | LCCN (EN) sh91003964 · GND (DE) 4157617-2 · J9U (EN, HE) 987007532217005171 |
|---|