
| Insonnia familiare fatale | |
|---|---|
| Malattia rara | |
| Specialità | psichiatria, medicina del sonno e neuropatologia |
| Eziologia | Genetica |
| Sede colpita | Sistema nervoso centrale |
| Classificazione e risorse esterne (EN) | |
| ICD-9-CM | 046.8046.8 |
| ICD-10 | A81.83 |
| OMIM | #600072 |
| MeSH | D034062 |
| Sinonimi | |
| FFI | |

L'insonnia familiare fatale (IFF) è una malattia genetica da prioni, autosomica dominante, che conduce alla degenerazione dei nuclei del talamo e della corteccia cerebrale. Il gene PRNP mutato codifica la proteina PrPSc, che tende a ripiegarsi in una maniera errata, detta forma prionica. L'insonnia fatale familiare è una malattia degenerativa del cervello appartenente al gruppo delle encefalopatie spongiformi.
Causa insonnia totale (impossibilità di dormire) ed evidenti sintomi neurologici e, dopo alcuni mesi di sofferenza, porta alla morte. Attualmente non esistono farmaci in grado di curare la malattia, tuttavia diversi studi sottolineano l'azione preventiva dell'antibiotico doxiciclina. Le persone che ne soffrono presentano alterazioni strutturali di particolari proteine, chiamate prioniche, resistenti alla degradazione che si accumulano nel cervello, in direzione del talamo, erodendolo e causando l'impossibilità di dormire, e di conseguenza il decesso.
Il primo caso di cui si ha notizia coinvolge un uomo italiano, morto a Venezia nel 1765.
Storia
La scoperta di questa patologia si deve all'osservazione del complesso e misterioso quadro sintomatologico mostrato da tre fratelli di una famiglia della provincia di Treviso, che si ammalarono consecutivamente a partire dal 1973. Al primo caso, una donna di 48 anni, fu diagnosticata la sindrome di Menière, ma, essendo risultata inutile ogni terapia, morì dopo 8 mesi dalla comparsa dei primi sintomi; neanche con l'esame autoptico si arrivò a una conclusione sulla causa del decesso. Stessa cosa avvenne con sua sorella, ammalatasi 5 anni dopo, poiché il neurologo Erwin Wildi non riuscì ad individuare la malattia letale attraverso l'esame di parte dell'encefalo. Pertanto proseguirono con maggior interesse gli studi sulla genealogia delle due pazienti e sui racconti dei loro familiari, anche se molti consanguinei erano emigrati nel corso del tempo.
Dopo un periodo di interruzione, si ritornò ad investigare, quando, nel 1984, venne ricoverato il fratello cinquantatreenne delle due pazienti, che, pur non avendo mai manifestato problemi di salute particolarmente gravi, cominciò a soffrire di insonnia, senza ricevere alcun beneficio dalla somministrazione di farmaci ipnoinducenti, tanto che morì dopo 9 mesi dall'esordio della malattia. I vari sintomi del terzo paziente vennero sistematicamente descritti da Lugaresi, medico in una clinica di Bologna. Dopo l'individuazione di 5 nuovi casi e approfonditi studi anatomopatologici, con cui si individuarono gli effetti della malattia nei nuclei anteriore e dorso-mediale del talamo, Ignazio Roiter, al quale si deve la prima descrizione clinica della malattia basata sull'osservazione dei tre fratelli, fondò l'Associazione Familiari Insonnia Familiare Fatale. Dalle approfondite ricerche sulla storia degli antenati dei pazienti emerse che il primo caso di portatore della malattia potrebbe essere rappresentato da un tale Giacomo, ricco medico veneziano deceduto, come si legge nei registri parrocchiali, nel 1765 dopo aver manifestato dei sintomi apparentemente inspiegabili, tra cui un'estenuante insonnia durata 9 mesi.
Epidemiologia
L'età media della malattia è intorno ai 40 anni, con una variabilità compresa tra i 20 e i 60 anni. Si tratta di una malattia rarissima, che in tutto il mondo è stata diagnosticata a circa 200 pazienti e coinvolge 27 famiglie. Casi sono stati documentati in Austria, Germania, Francia, Cina, Giappone e Stati Uniti d'America, mentre in Italia i pazienti affetti da questa patologia appartengono a due famiglie distinte.
Eziologia e patogenesi

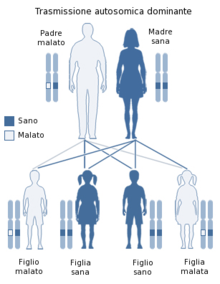
Le malattie da prioni sono causate dalla conversione di una proteina normale (PrPC o prion normale) in una forma ad essa simile (PrPSc), che, essendo insolubile e resistente agli enzimi, si accumula nel sistema nervoso danneggiandolo. In particolare, nel caso dell'insonnia familiare fatale la parte più colpita è quella del talamo (nuclei anteriore e dorso-mediale), ma vengono coinvolti anche l'oliva bulbare ed alcuni punti isolati della corteccia cerebrale. Nel cromosoma 20 è presente una mutazione genetica puntiforme del codone 178 del gene della PrP, che codifica asparagina al posto dell'acido aspartico, mentre il codone 129 dello stesso allele, che è interessato da un polimorfismo, la metionina. Di questa malattia a trasmissione dominante esistono due varianti legate al codone 129; infatti se il codone dell'altro cromosoma 20 codifica la valina, l'evoluzione del disturbo è più lunga, se invece il paziente è omozigote rispetto a questo codone, si ha un'evoluzione più breve e si assiste ad un minor danneggiamento della corteccia cerebrale.
Pare che il meccanismo di insorgenza della malattia sia legato ad alterazioni ormonali, tra cui l'aumento delle catecolamine e del cortisolo, mancanza di melatonina e perdita del ritmo circadiano dell'ipofisi. Inoltre, secondo alcuni studi scientifici, la proteina PrPc sarebbe una componente dei recettori del GABA e la sua conversione in un'isoforma proteica causerebbe l'incapacità degli stessi recettori di esercitare la loro azione inibitrice sulle cellule nervose del talamo, che, a causa dell'eccessiva attivazione, andrebbero incontro a necrosi.
Esiste anche una forma sporadica dell'Insonnia Familiare, definita sFI, in cui l'unica mutazione presente è al codone 129 ma senza la mutazione al codone 178.
Clinica
Segni e sintomi

Dopo un periodo di spossatezza, il paziente comincia a soffrire di insonnia farmacoresistente sempre più grave, fino all'incapacità completa di raggiungere il sonno profondo per poi presentare condizioni come lo stupor ed il coma. Tuttavia nello stadio in cui si manifesta l'impossibilità di dormire ci sono fasi di sonno paradosso apparentemente simili al sonnambulismo, che sono caratterizzate da sogni recitati, e questo la rende molto diversa dalla insonnia ordinaria. La malattia danneggia progressivamente la memoria verbale ed a lungo termine, ma non comporta demenza.
Oltre all'insonnia, si osservano difficoltà motorie (es. andatura atassica) e una precoce disautonomia neurovegetativa, con disturbi sfinterici, scialorrea, lacrimazione, sudorazione costante, rinorrea, incremento della temperatura corporea e della pressione arteriosa, tachicardia e alterazioni del respiro (es. tachipnea), ma talvolta compaiono anche paresi.
Trattamento
Nel marzo 1984, il neurologo italiano ed esperto del sonno Benedetto Ignazio Roiter ricevette un uomo di 53 anni presso l'ospedale di Treviso e per primo descrisse clinicamente la malattia. Il paziente, citato nei testi solo con il nome di Silvano, aveva infatti deciso in un raro momento di coscienza di farsi studiare dai medici e di donare alla ricerca il proprio cervello dopo la morte, nella speranza di trovare una cura per le future vittime. La ricerca venne pubblicata nel 1986 sul New England Journal of Medicine.
Al 2022, nessuna cura o trattamento è stato ancora trovato per la IFF, né la terapia genica (finanziata anche grazie al contributo di Telethon) finora ha avuto successo. Sebbene non sia attualmente possibile invertire la malattia sottostante, vi sono alcune prove che i trattamenti incentrati esclusivamente sui sintomi possono migliorare la qualità della vita.
Per la forma sporadica, esistono studi recenti relativi a derivati fenotiazinici, ma sono ancora preliminari, nonostante alcuni pazienti hanno stabilito che il sonno è migliorato con le fenotiazine originarie. In realtà però ci sono altri studi, come ad esempio gli studi sulla doxiciclina che sembrano promettenti in vitro che sono stati effettuati da un gruppo di scienziati Italiani.
Prognosi
La prognosi è sempre sfavorevole: la durata prima del decesso varia dai 7 ai 25 mesi, ma in media si aggira sui 13 mesi.
Bibliografia
- (EN) Sleep Medicine: Sleep Disorders, Fatal Familial Insomnia, Morvan's Syndrome, Sleep Apnea, Jet Lag, African Trypanosomiasis, Bruxism, General Books, 2010.
- (EN) Ronald Cohn e Jesse Russell, Fatal Familial Insomnia, Book on Demand Ltd., 2012.
- (EN) Christian Guilleminault, Fatal familial insomnia: inherited prion diseases, sleep, and the thalamus, Raven Press, 1994.
- (EN) Daniel T. Max, The Family That Couldn't Sleep: Unravelling a Venetian Medical Mystery, 2006, ISBN 1-4000-6245-4.
- (EN) Research Laboratories Merck, The Merck Manual quinta edizione, Milano, Springer-Verlag, 2008, ISBN 978-88-470-0707-9.
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su insonnia familiare fatale
Wikimedia Commons contiene immagini o altri file su insonnia familiare fatale
Collegamenti esterni
- AFIFF - Associazione Familiari Insonnia Familiare Fatale (Malattie da Prioni), su afiff.net.
- AIEnP - Associazione Italiana Encefalopatie da Prioni), su aienp.it.
- The Cellular Prion Protein (PrPC): Its Physiological Function and Role in Disease
| Controllo di autorità | BNF (FR) cb13549188z (data) |
|---|