
| Malattia di Hirschsprung | |
|---|---|
 | |
| Malattia rara | |
| Cod. esenz. SSN | RN0200 |
| Specialità | pediatria, digestive system surgery e gastroenterologia |
| Classificazione e risorse esterne (EN) | |
| OMIM | 600156, 606874, 606875, 608462 e 611644 |
| MeSH | D006627 |
| MedlinePlus | 001140 |
| eMedicine | 178493 |
| Sinonimi | |
| Megacolon congenito agangliare | |
| Eponimi | |
|
Harald Hirschsprung Giuseppe Mya Frederik Ruysch | |
La malattia di Hirschsprung (HSCR), o megacolon congenito agangliare, è una malattia congenita dell'intestino caratterizzata dall'assenza, per un tratto del canale alimentare, del plesso mioenterico e del plesso sottomucoso, che garantiscono la coordinazione dei movimenti peristaltici intestinali.
Deve il suo nome al medico danese Harald Hirschsprung che per primo la descrisse nel 1886. Tuttavia è nota anche come malattia di Mya, da Giuseppe Mya, e come malattia di Ruysch, da Frederik Ruysch.
Epidemiologia
Ha una incidenza di circa 1 caso ogni 5.000 nati vivi e dimostra un'evidente predilezione per il sesso maschile, con un rapporto tra maschi e femmine pari a 4:1. Nel sesso femminile la malattia è presente soprattutto a segmento lungo, ovvero coinvolge un ampio tratto di intestino, mentre nel maschio a segmento corto, ovvero limitato al retto-sigma. Il 10% dei casi si manifesta in pazienti con sindrome di Down.
Eziologia
Si tratta di una malattia multigenicamalattia genetica, osservata inizialmente in pazienti affetti da trisomia 21. Successivamente sono state dimostrate correlazioni con delezioni dei geni EDNRB, situato sul cromosoma 13 (-q22), RET, sul cromosoma 10 (-q11.2), e ZFHX1B, sul cromosoma 2 (-q22). Nel 70% delle forme lunghe di affezione è il gene coinvolto è RET.
È stato osservato che la mutazione alla base del morbo di Hirschsprung, a carico del gene RET, è di tipo loss-of-function; essa è correlata a una mutazione di tipo gain-of-function nel medesimo locus, che a sua volta provoca la patologia MEN2 (neoplasie endocrine multiple di tipo 2). Ciò è un tipico esempio di eterogeneità allelica clinica. Studi recenti dimostrano come possa esserci un'associazione tra anomalie congenite del rene e del tratto urinario (CAKUT) e la malattia di Hirschsprung.
La malattia è dovuta ad anomalie di sviluppo e maturazione del sistema nervoso enterico a partire dalle creste neurali. Ne consegue l'assenza di gangli dei plessi mioenterico di Auerbach e sottomucoso di Meissner per un'estensione variabile, che si manifesta clinicamente con un'assenza di peristalsi nel tratto agangliare.
È sempre interessato il retto e solitamente anche il colon sigmoideo. Meno frequentemente è interessato l'intero colon, mentre in casi del tutto eccezionali tutto l'intestino.
Clinica
Si manifesta in età molto precoce, generalmente nei primi giorni di vita, e si presenta quasi costantemente con ritardata emissione del meconio. È tuttavia generalmente riconosciuto che il quadro clinico di presentazione della malattia di Hirschsprung può risultare molto variabile per forma e severità. Spesso l'emissione di meconio risulta parziale, piuttosto che ritardata. Paradossalmente, diarrea cronica, associata a segni e sintomi di proctite, possono costituire la prima manifestazione.
Possono individuarsi quattro quadri clinici principali:
- occlusione neonatale persistente: ben presto l'accumulo di feci e gas nel colon dilatato, al di sopra del segmento agangliare, diviene estremo, e non è più possibile un'attività motoria compensatoria a carico del colon normale. Il quadro clinico è caratterizzato da assenza di evacuazioni spontanee, distensione addominale ingravescente e vomito. La sovradistensione del colon e il danno della parete possono tragicamente evolvere nell'embolismo gassoso del sistema portale. Questo quadro necessita di una precoce colostomia derivativa sul segmento di colon normogangliare (colostomia di livello);
- costipazione cronica progressiva: quando i segni iniziali, legati alla ritardata emissione di meconio, si risolvono spontaneamente o con la semplice stimolazione rettale, e subentra un quadro di stipsi cronica progressiva, che risponde ai normali clisteri di pulizia o a determinati lassativi. I pazienti di questo gruppo, quando correttamente diagnosticati, giungono all'intervento chirurgico radicale senza la necessità di eseguire un intervento colostomico d'urgenza;
- esordio sfumato con evoluzione ostruttiva: quando i pazienti presentano per settimane o mesi segni lievi di malattia, seguiti da un periodo di stipsi ostinata, che evolve in ostruzione cronica. Questi bambini manifestano una grave distensione gassosa dell'addome; all'impossibilità di emissione di gas per la sovradistensione colica, si associano aumentati processi di crescita batterica. È comune il riscontro di segni di distrofia generale con anoressia, episodi infettivi ricorrenti, eventuale anemia ipocromica e ipoproteinemia. Se non trattato chirurgicamente il quadro evolve verso gravi crisi subocclusive o verso l'enterocolite secondaria;
- enterocolite precoce secondaria: quando compare “diarrea paradossa”, con feci liquide, maleodoranti, miste a muco. Il quadro è il risultato delle alterazioni della mucosa colica legate alla stasi fecale, alla sovracrescita batterica, al reflusso fecale cieco-ileale. Questo tipo di presentazione non può trovare completa spiegazione nelle sole ragioni meccaniche, sono state infatti documentate complesse alterazioni immunitarie nella mucosa dei soggetti che manifestano enterocolite associata a HSCR. La perdita di liquidi ed elettroliti può evolvere verso l'acidosi metabolica, o persino morte per shock settico.
Diagnostica
La diagnosi della malattia di Hirschsprung non è difficile e solitamente anche la sola clinica può essere sufficiente; essa entra in diagnosi differenziale con la stipsi ostinata secondaria a fecaloma, ma se ne differenzia per l'età d'insorgenza. Nel primo caso questa si manifesta precocemente e, all'esplorazione rettale, l'ampolla risulta vuota; nel secondo caso l'insorgenza è verso i 4-5 anni e l'ampolla risulta piena di feci.
La diagnosi può essere effettuata tramite l'esecuzione di un clisma opaco dell'intestino, di una biopsia rettale e di una manometria anorettale, se si tratta di bambini più grandicelli o adulti.
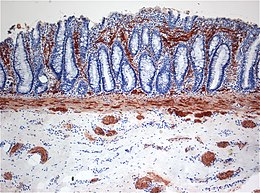
In particolare alla biopsia, che rappresenta lo standard di riferimento per la diagnosi, si osserva l'assenza di gangli nervosi nella tonaca muscolare e nella sottomucosa, associata a incremento di fibre nervose positive alla tecnica di colorazione della acetilcolinesterasi (vedi Illustrazione sezione bioptica colorata) . Quasi costantemente si assiste a dilatazione e ipertrofia del tratto di colon normale posizionato a monte di quello affetto. La mucosa può risultare infiammata e può ulcerarsi. La diagnosi di certezza è raggiunta, tuttavia, solo tramite colorazione per l'acetilcolinesterasi dei campioni di sottomucosa.
Trattamento
Il trattamento della malattia di Hirschsprung è chirurgico e prevede la rimozione del tratto agangliare di intestino, successivamente alla riduzione del diametro del megacolon a monte tramite lavaggi rettali o il confezionamento di una colostomia temporanea. Quest'ultima è preferibilmente da evitarsi, per via dell'alta incidenza di infezioni. L'intervento deve preferenzialmente essere effettuato il prima possibile, ovvero nei primi giorni-settimane di vita.
Sono state descritte tre tecniche chirurgiche principali:
- Tecnica di Swenson (1948): consiste nella resezione del tratto sprovvisto di gangli e dell'abbassamento del tratto a monte fino all'ano;
- Tecnica di Duhamel (1956): è simile alla precedente, ma prevede la conservazione del retto, dietro al quale viene abbassato il colon funzionale;
- Tecnica di Soave (1960): questo intervento, facilmente eseguibile anche per via laparoscopica, prevede la rimozione della mucosa intestinale e l'abbassamento, all'interno del retto, del colon funzionale.
Prognosi
La mortalità della malattia di Hirschsprung è solitamente inferiore al 3%, ma può raggiungere il 10% nei casi di enterocolite e nei casi in cui il tratto agangliare raggiunga l'ileo. Complicanza principale dell'intervento chirurgico può essere la costipazione post-operatoria persistente (7%-13% dei casi).
Bibliografia
- Research Laboratories Merck, The Merck Manual quinta edizione, Milano, Springer-Verlag, 2008, ISBN 978-88-470-0707-9.
- Robbins & Cotran, La basi patologiche delle malattie, Milano, Elsevier Italia, 2006, ISBN 88-85675-53-0.
- Harrison, Principi di medicina interna, 16ª ed., Milano, McGraw-Hill, 2005, ISBN 88-386-2999-4.
- Renzo Dionigi, Chirurgia, Milano, Masson - Elsevier, 2006, ISBN 88-214-2912-1.
Voci correlate
- Sindrome da tappo di meconio
- Enterocolite di Hirschsprung
- Atresia anale
- Megacolon
- Ipoganglionosi del colon
- Disganglionosi
- Desmosi intestinale
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su malattia di Hirschsprung
Wikimedia Commons contiene immagini o altri file su malattia di Hirschsprung
Collegamenti esterni
- (EN) Malattia di Hirschsprung, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.