
| Sindrome del QT breve | |
|---|---|
 | |
| Specialità | cardiologia e genetica clinica |
| Eziologia | mutazione |
| Classificazione e risorse esterne (EN) | |
| ICD-10 | R94.3 |
| OMIM | 609621, 609620 e 609622 |
| MeSH | C580439 |
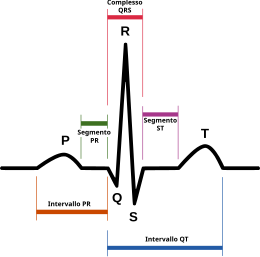
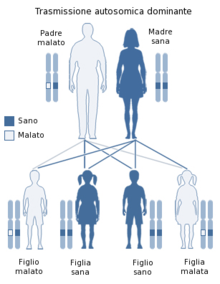
La sindrome del QT breve (abbreviazione dall'inglese SQTS, Short QT Syndrome) è una malattia genetica molto rara del sistema elettrico del cuore ed è associata ad un aumentato rischio di presentare anomalie del ritmo cardiaco e morte cardiaca improvvisa. È caratterizzata da un ampio spettro di segni e sintomi: un intervallo QT breve nell'elettrocardiogramma (ECG) (≤ 300 ms) che non cambia in maniera significativa con il ritmo cardiaco, alte e appuntite onde T, e un cuore strutturalmente sano. Essa è causata da mutazioni nei geni che codificano i canali ionici che accorciano il potenziale di azione cardiaca. La sindrome del QT breve è, quindi, una malattia congenita ed è ereditata come carattere autosomico dominante.
La condizione viene diagnosticata analizzando un tracciato ECG a 12 derivazioni e può essere trattata utilizzando un defibrillatore cardiaco impiantabile o grazie ad una terapia farmacologica, come la chinidina. La sindrome del QT corto è stata descritta per la prima volta nel 2000 e la prima mutazione genetica associata alla condizione è stata identificata nel 2004.
Storia
La prima segnalazione della sindrome del QT breve si trova in una pubblicazione del 2000, in cui era stata descritta una famiglia i cui componenti presentavano in giovane età, all'ECG a 12 derivazioni, una fibrillazione atriale e un paziente non correlato che aveva subito una morte cardiaca improvvisa associata a un intervallo QT breve. La correlazione tra un QT breve e la morte cardiaca improvvisa è stata descritta nel 2003 e il primo gene associato alla condizione è stato identificato nel 2004. Nel 2011 sono stati proposti criteri per la diagnosi.
Epidemiologia
La sindrome del QT corto è una condizione molto rara con, sl 2018, meno di 300 casi descritti nella letteratura medica. Essendo una malattia genetica, coloro che ne sono affetti sono nati con essa. Si può manifestare potenzialmente come una sindrome della morte improvvisa del lattante nei neonati. I maschi e le femmine hanno la stessa probabilità di presentare questa sindrome e hanno uno stesso rischio di morte cardiaca improvvisa.
Sintomi
Le persone affette da sindrome da QT breve presentano un rischio aumentato di sviluppare malattie cardiache. Anormali ritmi cardiaci si presentano negli infanti e possono assumere forme come la fibrillazione atriale, portando i pazienti a lamentare diversi sintomi come dispnea o affaticamento. Tutti coloro che ne soffrono, vanno ad incontro a sincopi (perdita di coscienza) apparentemente inspiegabili.
Il riscontrarsi di una fibrillazione atriale in età neonatale, pertanto, dovrebbe sollevare il sospetto di una sindrome del QT breve. Inoltre, possono verificarsi anche disturbi del ritmo cardiaco molto più pericolosi, come la fibrillazione ventricolare che porta alla morte cardiaca improvvisa. Più di un terzo di coloro con un QT breve, presenta aritmie ventricolari o morte cardiaca improvvisa. I casi vengono rilevati nei neonati durante lo screening familiare o casualmente nell’eseguire un elettrocardiogramma (ECG) per un altro motivo.
Diagnosi
La diagnosi della Sindrome del QT breve consiste nell'analisi della storia clinica, nella lettura del tracciato ECG e nello studio elettrofisiologico endocavitario. Attualmente non esistono linee guida per la diagnosi di questa patologia in quanto solo poche decine di casi sono stati riportati a livello mondiale.
Elettrocardiogramma a 12 derivazioni

La caratteristica della Sindrome del QT breve in un tracciato ECG a 12 derivazioni è appunto un intervallo QT breve, che corrisponde ad un QTc inferiore ai 330 ms, con un cuore strutturalmente sano. Infatti, normalmente l'intervallo QT varia con la frequenza cardiaca: in condizioni normali diminuisce all’aumentare dei battiti. Questa variazione però non avviene nei soggetti con sindrome del QT breve. Si raccomanda, pertanto, di valutare l'intervallo QT a una frequenza cardiaca prossima a 60-70 battiti al minuto. Inoltre, sempre all'analisi del tracciato ECG, si possono notare onde T alte e strette con depressione del segmento PR. Alcuni individui possono anche presentare fibrillazione atriale sottostante.
Studi elettrofisiologici
Allo studio elettrofisiologico i soggetti affetti da SQTS manifestano periodi refrattari brevi, sia a livello atriale così come a livello ventricolare. Inoltre, con la stimolazione elettrica programmata, viene spesso indotta fibrillazione ventricolare.
Eziologia
L'eziologia della Sindrome del QT breve non è ancora chiara al momento. Una delle ipotesi ad oggi è che sia causata dall'aumento dell'attività delle correnti di potassio in uscita, nella fase 2 e 3 del potenziale d'azione. Questo provocherebbe un accorciamento della fase di plateau (fase 2), abbreviando quindi l'intero potenziale d'azione e portando ad una complessiva riduzione dei periodi refrattari e dell'intervallo QT. Nelle famiglie affette da SQTS sono state descritte due differenti mutazioni missens nel gene HERG (human ether-a-go-go). Queste mutazioni determinano la sostituzione dello stesso amminoacido nel canale ionico IKr. Il canale IKr mutato presenta un incremento dell'attività rispetto ad un canale normale. Una lista di geni le cui varianti sono state associate con la sindrome del QT breve può essere trovata qui sotto:
| Tipo | OMIM | Gene | Note |
|---|---|---|---|
| SQT1 | 609620 | KCNH2 | Conosciuto anche come hERG, codifica il canale potassio KV11.1 responsabile per la corrente rettificante ritardata IKr |
| SQT2 | 609621 | KCNQ1 | Codifica il canale potassio responsabile per la corrente di potassio rettificante ritardata IKs |
| SQT3 | 609622 | KCNJ2 | Codifica per il canale potassio Kir2.1 responsabile per la corrente di potassio rettificante interna IK1 |
| SQT4 | 114205 | CACNA1C | Codifica per la subunità alfa del trasportatore del canale del calcio di tipo L ICa(L) |
| SQT5 | 114204 | CACNA2D1 | Codifica la subunità alfa2/delta del trasportatore del canale del calcio di tipo L ICa(L) |
| SQT6 | 106195 | SLC4A3 | Codifica lo scambiatore bicarbonato / cloro |
Fisiopatologia
Le mutazioni nei geni KCNH2, KCNJ2 e KCNQ1 sono la causa della Sindrome del QT breve. Questi geni codificano proteine-canale di membrana. Questi canali permettono il passaggio con carica positiva (ioni) di potassio dall'interno all'esterno delle cellule e viceversa. Nel muscolo cardiaco, questi canali ionici hanno un ruolo fondamentale nel mantenimento del ritmo normale del cuore.
Le mutazioni nei geni KCNH2, KCNJ2 e KCNQ1 incrementano l'attività dei canali, che cambiano il flusso degli ioni di potassio tra le cellule. Questa alterazione nel trasporto ionico modifica la maniera nella quale batte il cuore, caratterizzando un ritmo cardiaco anormale, tipico della Sindrome del QT breve.
A causa dell'ereditarietà a carattere autosomico dominante, la maggior parte dei soggetti avranno una storia in famiglia di morte improvvisa in età giovanile (anche in infanzia), palpitazioni, o fibrillazione atriale. La SQTS è associata ad un incremento del rischio di morte cardiaca improvvisa, per la maggior parte a causa di fibrillazione ventricolare.
Trattamento
Il trattamento per la sindrome del QT corto mira a prevenire ritmi cardiaci anormali e a ridurre il rischio di incorrere in una morte cardiaca improvvisa. Poiché la condizione è molto rara, è stato difficile testare potenziali trattamenti sperimentali, quindi l'eventuale efficacia è stata valutata empiricamente in base al consenso della comunità scientifica. Oltre a trattare l'individuo ritenuto affetto dalla condizione, si consiglia di sottoporre i suoi familiari ad uno screening.
Defibrillatore cardiaco impiantabile

Ai soggetti con sindrome del QT corto che hanno già accusato un anormale ritmo cardiaco potenzialmente letale, come la fibrillazione ventricolare, può essere raccomandato un defibrillatore cardiaco impiantabile(ICD) al fine di ridurre la probabilità di una morte improvvisa. Questo dispositivo viene impiantato sotto la cute ed è in grado di monitorare continuamente il ritmo cardiaco. Se il dispositivo rileva un pericoloso disturbo di esso, rilascia una piccola scossa elettrica con lo scopo di ripristinare un ritmo normale. L'opportunità dell'impianto di un ICD nei soggetti con sindrome del QT corto che non hanno ancora avuto un'aritmia potenzialmente letale è controversa, ma questa misura può comunque essere presa in considerazione.
Trattamento farmacologico

È stato dimostrato che la chinidina, un agente antiaritmico di classe IA, sia in grado di correggere parzialmente l'intervallo QT per il suo effetto nel prolungare la durata del potenziale d'azione. Al momento l'efficacia a lungo termine di questi farmaci non è dimostrata ma si possono rivelare utili in associazione con un defibrillatore per ridurre il numero di eventi aritmici. Il sotalolo, un altro antiaritmico, può prolungare il QT in alcuni sottotipi di sindrome del QT breve. Sono stati provati altri farmaci, inclusi beta-bloccanti, flecainide e amiodarone, ma al momento vi sono poche prove a sostegno del loro uso.
I farmaci possono anche essere usati per trattare la fibrillazione atriale, un ritmo cardiaco anormale meno pericoloso e anch'esso associato a un breve QT. Anche il propafenone, un antiaritmico di classe IC, può essere utile per prevenire la fibrillazione atriale. Coloro che sviluppano una fibrillazione atriale possono anche richiedere farmaci per ridurre la coagulazione del sangue al fine di ridurre il rischio di ictus.
Prognosi
La rarità della sindrome del QT breve rende estremamente difficile formulare una prognosi precisa. È stato stimato che il rischio di morte cardiaca improvvisa si attesti intorno allo 0,8% all'anno, portando a un rischio cumulativo di morte cardiaca improvvisa del 41% entro i 40 anni. Essere incorsi precedentemente in un arresto cardiaco, comporta una maggiore probabilità di ulteriori aritmie pericolose. Alcuni studi hanno suggerito che coloro che presentano intervalli QT più brevi potrebbero avere un rischio più elevato di sviluppare aritmie, ma non tutti concordano con ciò. I risultati degli studi elettrofisiologici invasivi non sono in grado di predire quale sia il rischio di arresto cardiaco in un individuo con sindrome del QT corto.
Bibliografia
- Braunwald, Fauci, Kasper, Hauser, Longo, Jameson, Loscalzo, Harrison - Principi di Medicina Interna, Milano, Elsevier Masson, 2009, ISBN 978-88-214-2987-3.
- Eugene Braunwald, Malattie del cuore (7ª edizione), Milano, McGraw-Hill, 2007, ISBN 978-88-386-3940-1.
- Hurst, Il Cuore (il manuale - 11ª edizione), Milano, McGraw-Hill, 2006, ISBN 978-88-386-2388-2.
- (EN) Lilly L.S., Pathophysiology of Heart Disease., 2007ª ed., Baltimore: Lippincott Williams & Wilkins, ISBN 978-1-60547-723-7.
- Rowlands DJ, Interpretazione dell'elettrocardiogramma, 2004ª ed., Pro.Med. Editore, ISBN 978-88-6521-011-6.
Voci correlate
Collegamenti esterni
- National Library of Medicine. Short QT syndrome