
| Sindrome di Down | |
|---|---|
 | |
| Malattia rara | |
| Cod. esenz. SSN | RN0660 |
| Specialità | genetica clinica e neurologia |
| Eziologia | Genetica |
| Classificazione e risorse esterne (EN) | |
| OMIM | 190685 |
| MeSH | D004314 |
| MedlinePlus | 000997 |
| eMedicine | 943216 |
| Sinonimi | |
| Mongoloidismo Trisomia 21 | |
| Eponimi | |
| John Langdon Down | |
 | |
La sindrome di Down, più propriamente trisomia 21 e, in passato, mongolismo o mongoloidismo, è una condizione cromosomica causata dalla presenza di una terza copia (o una sua parte) del cromosoma 21. Viene anche abbreviata DS dall'acronimo del suo nome in inglese, Down syndrome. Si tratta della più comune anomalia cromosomica del genere umano, solitamente associata a un ritardo nella capacità cognitiva e nella crescita fisica, oltre che a un particolare insieme di caratteristiche del viso.
Il QI medio degli individui con la sindrome di Down è circa 50, contro il 100 delle persone non affette. Laddove tutti i casi diagnosticati presentano un ritardo cognitivo, la disabilità è molto variabile tra gli individui affetti. La maggior parte rientra nella gamma di «poco» o «moderatamente disabili» dal punto di vista della capacità motoria.
La sindrome prende il nome dal medico britannico John Langdon Down, che ne fece un'ampia descrizione nel suo Observations on An Ethnic Classification of Idiots (1866) benché vi siano al riguardo studi clinici precedenti a opera dei medici francesi J.E.D. Esquirol (1838) ed Édouard Séguin (1844). Nel 1958 ne fu identificata la causa genetica in una trisomia del cromosoma 21.
La sindrome di Down può essere identificata in un bambino anche prima della nascita con lo screening prenatale.
Storia

Il primo medico a caratterizzare la condizione quale forma distinta di disabilità mentale fu John L. Down nel 1862, che la descrisse più ampiamente nel 1866, con il testo citato. Down coniò il termine «mongoloide» per descrivere le persone affette dalla sindrome, in ragione della similitudine somatica con i bambini di etnia "mongola", secondo la terminologia risalente al lavoro di Blumenbach (1795), allora in uso ma in seguito abbandonata.
Quasi un secolo più tardi, nel 1961, diciannove genetisti scrissero una lettera al direttore della rivista scientifica britannica Lancet, richiamando l'attenzione sul fatto che detto termine potesse avere «connotazioni fuorvianti» e che fosse ormai una «locuzione imbarazzante» da abbandonare. La rivista britannica promosse e sostenne quindi l'adozione del termine sostitutivo «sindrome di Down».
Parimenti i termini «mongolismo» o «imbecillità mongola» caddero progressivamente in disuso fin dall'inizio degli anni settanta e oggi sono considerati inaccettabili, svilenti o offensivi. L'Organizzazione mondiale della sanità non usa più il termine ufficialmente dal 1965 a seguito di una richiesta formale del delegato della Repubblica Popolare Mongola. I gruppi di tutela dei genitori dei bambini affetti dalla sindrome accolsero favorevolmente l'eliminazione della definizione di «mongoloide», ritenuta impropria e discriminatoria.
Nel 1975 i National Institutes of Health degli USA convocarono una conferenza per standardizzare la denominazione. Il termine trisomia 21 è sinonimo ormai consolidato.
Nel corso del XX secolo la sindrome di Down divenne la forma più riconoscibile di disabilità mentale. La maggior parte degli individui affetti iniziò a ottenere cure mediche riguardo ai problemi associati. Con la nascita del movimento eugenetico, in 33 degli allora 48 stati statunitensi e in diversi altri paesi iniziarono programmi di sterilizzazione forzata delle persone con sindrome di Down e con gradi di disabilità comparabili; per esempio Aktion T4 nel Terzo Reich, programma di sterminio sistematico.
Fino alla metà del XX secolo la causa della sindrome di Down rimase sconosciuta; tuttavia era stata rilevata un'associazione con l'età materna. I testi medici riportavano tale condizione quale combinazione di fattori ereditari non ancora identificati. Altre teorie erano incentrate su possibili lesioni subite durante il parto.
Dal 1950, con lo sviluppo delle tecniche per analizzare il cariotipo, fu possibile identificare le anomalie cromosomiche di numero o di forma e nel 1958 fu individuata come causa della sindrome di Down la presenza di un cromosoma soprannumerario, per la prima volta pubblicato in un articolo di Marthe Gautier, Jérôme Lejeune e Raymond Turpin, e, di conseguenza, la condizione venne definita «trisomia 21».
Epidemiologia

Il fattore che maggiormente influenza l'incidenza della sindrome di Down è l'età materna. Come si nota dal grafico a destra, quando una donna supera i circa 35 anni di età la probabilità di concepire un figlio affetto da tale condizione aumenta considerevolmente. Tuttavia non si deve ritenere che i bambini che presentano la sindrome abbiano per la maggior parte madri anziane; infatti solo circa 1 su cinque ha una madre di età superiore ai 35 anni, questo perché la maggior parte delle donne concepisce la propria prole in età inferiore.
Secondo l'organizzazione mondiale della sanità l'incidenza della sindrome è compresa nell'intervallo 0,9 ÷ 1 caso ogni 1000 individui nati vivi. Negli Stati Uniti, il centro per la prevenzione e il controllo delle malattie fornisce una stima più elevata di questo dato con circa 1 bambino su 691 che nasce con la condizione. Se tuttavia si dovessero prendere in considerazione i concepimenti con trisomia 21, questi dati sarebbero molto più elevati poiché circa il 75% di queste gravidanze si concludono con un aborto o con la nascita di un feto privo di vita.
Uno studio effettuato nel Regno Unito, che ha analizzato dei dati tra il 1985 e il 2004, ha dimostrato un incremento della prevalenza della condizione sulle gravidanze e sulle nascite totali (da 1,3 a 2,5 su 1000), tuttavia non si è riscontrato alcun aumento considerando solo i nati vivi.
Il primo studio epidemiologico attendibile sulla sindrome di Down nella regione dell'Africa subsahariana risale al 1982 ed è stato effettuato in un ospedale in Nigeria. Questo studio ha stimato una prevalenza di 1,6 casi su 1000 nati vivi, in linea con i dati dei paesi occidentali. Successive indagini hanno portato a confermare o a sovrastimare, seppur di poco, questi valori. Esistono pochi dati sulla prevalenza della sindrome di Down nella popolazione africana, tuttavia un'analisi effettuata presso le zone rurali della regione Mpumalanga in Sudafrica ha messo in evidenza come si fossero registrati 2 soli casi di individui affetti dalla condizione su 4168 bambini tra i 2 e i 9 anni. Questo basso valore (1 su 2084) fa supporre come possa essere alta la mortalità nei primi mesi di vita per un bambino affetto da trisomia 21.
Se a livello globale i dati epidemiologici sulla sindrome di Down sono sovrapponibili e non risentono di correlazioni geografiche, vi è tuttavia l'eccezione degli Emirati Arabi, dove uno studio effettuato a Dubai, che ha preso in considerazione oltre 63.000 neonati tra il 1999 e il 2003, ha stimato un'incidenza della condizione di 1 ogni 449 nati vivi, con un tasso tra i cittadini di uno ogni 319, mentre una statistica più recente ha stimato un caso ogni 374 nati vivi, ovvero 267 affetti dalla condizione ogni 10.000 nascite. Ciò si può spiegare con la pressione che molte donne subiscono nel dover mettere al mondo più figli possibili e ciò le spinge quindi a concepirne in età avanzata. Lo stesso governo e gli usi locali favoriscono le donne che decidono di avere una prole numerosa.
Eziologia
L'età materna influenza le probabilità di concepire un bambino con sindrome di Down: uno studio condotto tra il 1970 e il 1989 sugli abitanti dell'Ohio e dell'area metropolitana di Atlanta ha mostrato che quando l'età della madre è compresa tra 20 e 24 anni la probabilità è 1 su 1562, tra 35 e 39 anni è 1 su 214, mentre sopra i 45 anni si attesta a 1 su 19. Sebbene le probabilità aumentino con l'età materna, l'80% dei bambini con sindrome di Down nasce da donne di età inferiore ai 35 anni: ciò è dovuto alla fecondità complessiva in tale fascia di età. Dati recenti suggeriscono inoltre che l'età paterna, in particolare quando supera i 42 anni, possa aumentare il rischio che la sindrome si manifesti nel bambino.
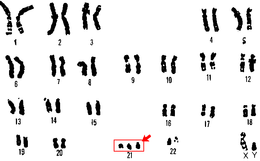
Nel 1958 fu dimostrato che la sindrome di Down è causata dalla presenza di un cromosoma 21 (o parte di esso) in più nel proprio patrimonio genetico, da qui la definizione di "trisomia 21", come sinonimo della sindrome stessa. In generale, ciò comporta una iperespressione di alcuni geni. Si stima che il cromosoma 21 contenga oltre 300 geni e una recente ricerca ha identificato la regione del cromosoma che contiene quelli principalmente responsabili delle manifestazioni cliniche della sindrome di Down. L'individuazione dei geni coinvolti può aiutare a indirizzare le cure mediche per le persone affette dalla condizione.
La presenza del materiale cromosomico in eccesso può avvenire in molti modi diversi: un cariotipo umano tipico è designato come 46,XX o 46,XY; ciò indica 46 cromosomi con due cromosomi sessuali XX tipico delle femmine e 46 cromosomi con due cromosomi sessuali XY che si riscontra nei maschi. Nell'1-2% dei casi, alcune delle cellule del corpo sono normali e altre cellule presentano la trisomia 21. Questa condizione viene chiamata sindrome di Down con mosaicismo (46, XX/47, XX, 21).
Trisomia 21
La trisomia 21 (conosciuta anche come cariotipo 47, XX,+21 per le femmine e 47,XY,+21 per i maschi), responsabile per circa il 95% dei casi di sindrome di Down, è causata da un evento meiotico non-disgiunzionale che si verifica in un gamete (uno spermatozoo o una cellula uovo) nel corso della meiosi, quando non si ha la separazione dei cromosomi omologhi in anafase I, o se non si verifica nel corso della meiosi II la separazione dei cromatidi fratelli. Come conseguenza, il gamete presenterà una copia extra del cromosoma 21, con un totale di 24 cromosomi. Se combinato con una cellula normale dall'altro genitore, l'embrione avrà quindi 47 cromosomi, con tre copie del cromosoma 21. Circa l'88% dei casi di trisomia 21 sono il risultato dalla non-disgiunzione nel gamete materno e l'8% da quello del gamete paterno, mentre nel 3% essa si verifica dopo che l'ovulo è stato fecondato dallo spermatozoo.
Traslocazione robertsoniana
Il materiale genico supplementare del cromosoma 21 che causa la sindrome di Down può essere dovuto a una traslocazione robertsoniana nel cariotipo di uno dei genitori. In questo caso, il braccio lungo del cromosoma 21 si fonde a un altro cromosoma acrocentrico (cioè caratterizzato da un centromero posto all'estremità), spesso il cromosoma 14 [45,XX o XY,t(14;21)(q10;q10)]. Una persona con una traslocazione è fenotipicamente normale. Durante la riproduzione, si ha un'alta probabilità di creare un gamete con un cromosoma 21 soprannumerario e quindi la nascita di un bambino con sindrome di Down. La condizione dovuta alla traslocazione è spesso definita come sindrome di Down familiare, è indipendente dall'età della madre ed è la causa del circa 4% dei casi osservati.
Mosaicismo
Una forma molto meno frequente di trisomia 21, definita "mosaicismo", si verifica circa nel 2% dei casi. Questa mutazione si presenta dopo il concepimento e la trisomia non si presenta in tutte le cellule dell'individuo ma solo in quelle che provengono dalla riproduzione della cellula mutata. La percentuale delle cellule colpite può variare da poche a quasi tutte, a seconda di quando si è verificata la segregazione anomala dei cromosomi omologhi.
Uno studio ha evidenziato che i casi di mosaicismo sono più comuni nelle femmine che nei maschi e non sembra esserci una correlazione con l'età materna.
Patogenesi
Il materiale genetico extra, presente nelle persone con sindrome di Down, comporta una sovraespressione di una parte degli oltre 300 geni localizzati sul cromosoma 21 ed è stata stimata intorno al 50%. Alcuni studi hanno suggerito che la regione critica per la sindrome di Down si trovi sulla regione 21q22.1-q22.3 del cromosoma, un'area che include i geni per la proteina precorritrice della beta-amiloide, l'enzima superossido dismutasi e probabilmente il proto-oncogene ETS2. Altre ricerche, tuttavia, non hanno confermato queste conclusioni.
Il ritardo mentale che si verifica nella sindrome di Down è dovuto ad un eccesso di betamiloide prodotto nel cervello, similmente alla malattia di Alzheimer. Questo peptide è elaborato dalla proteina precursore della beta-amiloide, il cui gene è localizzato sul cromosoma 21.Placche senili e ammassi neurofibrillari sono inoltre presenti in quasi tutti gli affetti a partire dai 35 anni, anche se una possibile demenza può non presentarsi. Coloro che sono affetti da Sindrome di Down mostrano anche una carenza nel normale numero di linfociti e producono un quantitativo inferiore di anticorpi e ciò contribuisce ad aumentare il rischio di incorrere in infezioni.
Espressione dell'eccesso di materiale genetico
La presenza di materiale genetico in eccesso negli affetti da trisomia 21 si traduce in una espressione biochimica maggiore di enzimi diversi di cui uno dei più popolari e più importanti è l'enzima superossido dismutasi (SOD, codificato dal gene 1) che gioca un ruolo fondamentale nella produzione di perossido di idrogeno (H2O2). Il suo livello superiore al normale porta a un'alterazione del metabolismo dell'ossigeno e una perossidazione maggiore dei lipidi e proteine con conseguente danno al DNA. Tutto ciò potrebbe essere causa sia dell'invecchiamento precoce sia della demenza presenile.
Gli altri geni, la cui espressione risulta aumentata, coinvolti nell'insorgenza dei disturbi associati con la sindrome di Down, sono:
- COL6A1: associato ai difetti cardiaci.
- ETS2: causa disturbi muscoloscheletrici.
- CAF1A: può interferire con la sintesi del DNA.
- CBS (Cistationina Beta Sintasi): può causare alterazione dei processi metabolici e di riparazione del DNA.
- DYRK: sembra essere correlato all'origine della disabilità cognitiva.
- CRYA1: può causare cataratta.
- GART: alterazione dei processi di sintesi e di riparazione del DNA.
- IFNAR: un gene coinvolto nella sintesi dell'interferone, il suo eccesso può provocare alterazioni nel sistema immunitario.
Segni e sintomi


I segni ed i sintomi della sindrome di Down sono caratterizzati dalla neotenia del cervello e del corpo allo stato fetale, dalla morfogenesi incompleta (vestigia) e da atavismo.
Gli individui con la sindrome possono avere alcune o tutte le seguenti caratteristiche fisiche: microgenia (mento anormalmente piccolo), fessure degli occhi oblique con pieghe della pelle all'angolo interno degli occhi (caratteristica già nota come "plica mongolica"),ipotonia muscolare (scarso tono muscolare), ponte nasale piatto, un'unica piega palmare, lingua sporgente (a causa della piccola cavità orale) e allargata vicino alle tonsille o macroglossia, viso piatto e largo, collo corto, macchie bianche su occhio e iride, note come macchie di Brushfield, eccessiva lassità articolare, spazio eccessivo tra alluce e il secondo dito, e dita corte.
I parametri di crescita, come altezza, peso e circonferenza della testa sono inferiori nei bambini con la condizione rispetto ai loro coetanei. Gli adulti con la sindrome di Down tendono ad avere bassa statura e gambe storte. L'altezza media per gli uomini è di 154 cm mentre per le donne è di 144 cm. Gli individui con sindrome di Down presentano un alto rischio di obesità.
| Caratteristica | Percentuale | Caratteristica | Percentuale |
|---|---|---|---|
| Bassa statura | 100% | Denti piccoli | 60% |
| Impronte digitali atipiche | 90% | Naso appiattito | 60% |
| Separazione dei muscoli addominali | 80% | Clinodattilia | 52% |
| Legamenti flessibili | 80% | Ernia ombelicale | 51% |
| Ipotonia | 80% | Collo corto | 50% |
| Brachicefalia | 75% | Mani corte | 50% |
| Genitali piccoli | 75% | Malattie cardiache congenite | 45% |
| Palpebre più grandi | 75% | Singola piega trasversale palmare | 45% |
| Estremità più corte | 70% | Macroglossia | 43% |
| Palato ovale | 69% | Volte epicantali | 42% |
| Orecchie basse e arrotondate | 60% | Strabismo | 40% |
| Macchie di Brushfield | 35% |

Le persone con sindrome di Down hanno un rischio più elevato di incorrere in diverse condizioni patologiche. Le conseguenze mediche provocate dal materiale genetico sovrannumerario sono molto variabili e possono influenzare la funzionalità di qualsiasi organo o di funzione fisiologica dell'organismo e ciò può contribuire a una minore aspettativa di vita. Tuttavia, a seguito dei miglioramenti nelle cure mediche, in particolare nei problemi cardiaci, la speranza di vita tra le persone con sindrome di Down è aumentata dai 12 anni che si potevano mediamente raggiungere nel 1912 ai 60 anni dei giorni nostri.
Caratteristiche mentali e neurologia
La maggior parte delle persone con sindrome di Down, stimabile nel 99,8%, presenta una disabilità intellettiva che va da lieve (QI 50 ÷ 70) a moderata (QI 35 ÷ 50), laddove il QI degli affetti da mosaicismo risulta mediamente più alto di 10 ÷ 30 punti di quello degli individui con trisomia 21. La metodologia del test di intelligenza è stata criticata per non aver preso in considerazione la correlazione con le disabilità fisiche, come ad esempio l'udito e i disturbi della vista che rallentano le prestazioni.
Comunemente gli individui con sindrome di Down hanno una buona comprensione del linguaggio, ma mostrano un ritardo nell'esprimersi verbalmente. Anche le abilità motorie mostrano dei ritardi e questo può interferire sullo sviluppo cognitivo del bambino.

Tuttavia le condizioni motorie sono molto differenti tra individuo e individuo. Alcuni bambini iniziano a camminare a circa 2 anni di età, mentre altri non camminano fino a quattro anni. La fisioterapia e/o la partecipazione a un programma di educazione fisica specifica può favorire lo sviluppo delle abilità motorie nei bambini con sindrome di Down.
I bambini e gli adulti con sindrome di Down presentano un rischio più elevato di sviluppare la sindrome di West, l'epilessia e la malattia di Alzheimer.
Malattia cardiaca congenita

L'incidenza delle cardiopatie congenite nei neonati con sindrome di Down arriva fino al 50% e il 7% di tutti i bambini con una cardiopatia risulta affetto dalla condizione.
Un difetto interventricolare è la forma più comune, con il 40% dei pazienti affetti. A seguire il difetto interatriale, riscontrabile nell'8%, la pervietà del dotto di Botallo, nel 7%, e infine l'1% dei nati con sindrome di Down presenta la tetralogia di Fallot.
Queste cardiopatie vengono, generalmente, diagnosticate precocemente, poiché mostrano fin dall'inizio alcuni segni e sintomi come un ritardo nella crescita, difficoltà respiratoria con tachipnea, tachicardia, cardiomegalia (aumento della dimensione del cuore), riduzione della diuresi e epatomegalia.
Queste malformazioni cardiache possono essere corrette chirurgicamente. Se ciò avviene in tempi stretti, il bambino non andrà incontro a complicazioni, specialmente a livello respiratorio, e potrà godere di una aspettativa di vita pari a quella degli altri bambini con sindrome di Down che non hanno presentato cardiopatie alla nascita.
Neoplasie
Anche se l'incidenza generale di neoplasie tra gli individui con sindrome di Down è la stessa che nel resto della popolazione, vi è una minor probabilità di sviluppare tumori maligni comuni, fatta eccezione per la leucemia e il cancro ai testicoli.
Le neoplasie ematologiche, come la leucemia, sono infatti più comuni nei bambini con sindrome di Down. In particolare, la leucemia linfoblastica acuta è almeno 20 volte più frequente nei bambini con la sindrome di Down e la forma megacarioblastica lo è di almeno 500 volte. La leucemia transitoria è una forma di leucemia rara negli individui senza sindrome di Down, ma colpisce fino al 20 % dei neonati che presentano la trisomia 21. Questa forma di leucemia è in genere benigna e si risolve da sola entro alcuni mesi, anche se potrebbe portare ad altre gravi patologie. In contrasto con le neoplasie ematologiche, i tumori maligni sono meno comuni, probabilmente a causa di un elevato numero di geni oncosoppressori contenuti nel materiale genetico extra.
Malattie della tiroide
Gli individui con sindrome di Down hanno un aumentato rischio di disfunzione alla tiroide, organo che collabora al controllo del metabolismo. L'ipotiroidismo è la condizione più comune, verificandosi in quasi un terzo dei pazienti. Ciò può essere causato dall'assenza della tiroide alla nascita (ipotiroidismo congenito) o per attacco autoimmune alla tiroide.
Malattie gastrointestinali
La sindrome di Down aumenta il rischio di incorrere nella malattia di Hirschsprung, dovuta alla mancanza di cellule nervose che controllano la funzione di parti del colon. Altre anomalie congenite che si verificano più frequentemente includono la stenosi ipertrofica del piloro, l'atresia duodenale e quella anale. La malattia da reflusso gastroesofageo e la celiachia sono inoltre più comuni tra le persone con sindrome di Down.
Infertilità
Le donne con sindrome di Down sono più fertili degli uomini ma hanno spesso difficoltà nella gravidanza, con aborti spontanei e parti prematuri. Senza diagnosi genetica preimpianto, circa la metà della prole di una persona con sindrome di Down presenterà la stessa condizione genetica. Gli uomini con sindrome di Down sono quasi uniformemente sterili, a causa di difetti della spermatogenesi. Vi sono stati solo tre casi documentati di maschi con la sindrome che hanno avuto figli.
Disturbi della vista e dell'udito

Disturbi della vista sono più frequenti negli individui con sindrome di Down. Quasi la metà di essi soffre di strabismo, e patologie che richiedono l'uso di occhiali sono altresì comuni. La cataratta (opacità del cristallino), il cheratocono e il glaucoma (pressione aumentata dell'occhio) sono condizioni che si verificano più comunemente negli individui con trisomia 21. Possono, inoltre, essere presenti le macchie di Brushfield (piccole macchie bianche o grigio/marrone sulla periferia dell'iride).
In generale, l'indebolimento dell'udito e le patologie legate ad esso si riscontrano nel 38 ÷ 78% dei bambini con sindrome di Down, rispetto al 2,5% dei bambini non affetti. Tuttavia, la diagnosi precoce e un trattamento aggressivo della malattia cronica dell'orecchio cronico possono portare circa il 98% dei bambini a livelli acustici normali.
Ogni componente del sistema uditivo è potenzialmente colpita dalla sindrome di Down. L'otite media con effusione è la causa più comune della perdita dell'udito nei bambini con tale condizione. L'infezione può iniziare alla nascita e cronicizzarsi durante la vita del bambino. Le infezioni all'orecchio sono principalmente associate con la disfunzione della tuba di Eustachio, a causa di alterazioni nella base cranica. Inoltre, un eccessivo accumulo di cerume può causare l'ostruzione del condotto uditivo esterno, spesso di dimensione ridotte nei bambini con la sindrome. La perdita dell'udito può variare da soggetto a soggetto, ma spesso, anche una lieve perdita può portare a gravi conseguenze per la percezione e l'acquisizione del linguaggio, se non diagnosticata e corretta in tempo.
Screening
Le linee guida raccomandano che lo screening per la sindrome di Down possa essere offerto a tutte le donne in stato di gravidanza, indipendentemente dall'età. Un certo numero di test possono essere utilizzati con diversi livelli di accuratezza. Essi sono di solito usati in combinazione per aumentare l'affidabilità del rilevamento, pur mantenendo un basso tasso di falsi positivi. Nessun test comunque può essere considerato definitivo, quindi se lo screening è positivo è necessario procedere all'amniocentesi o al prelievo dei villi coriali per confermare il sospetto. È consigliato effettuare i test di screening sia nel primo che nel secondo trimestre. Le diverse tecniche di screening in uso sono in grado di identificare fra il 90 e il 95% dei casi, con un tasso di falsi positivi compreso fra il 2 e il 5%.
| Test | Età gestazionale quando eseguito | Probabilità di rilevamento | Falsi positivi | Descrizione |
|---|---|---|---|---|
| Test combinato | 10-13.5 settimane | 82-87% | 5% | Utilizza l'ecografia per misurare la translucenza nucale (NT) oltre ad esami del sangue per i valori totali o liberi di beta-hCG e PAPP-A. |
| Tri test | 15-20 settimane | 81% | 5% | Misura nel siero materno i valori di alfa-fetoproteina, estriolo non coniugato, hCG, e inibina-A |
| Test integrato | 15-20 settimane | 94-96% | 5% | In combinazione con il tri test, il PAPP-A, e NT |
| DNA fetale libero circolante | Da 10 settimane | 96-100% | 0.3% | Un campione di sangue viene prelevato dalla madre mediante venipuntura e viene inviato per l'analisi del DNA. |
Ecografia
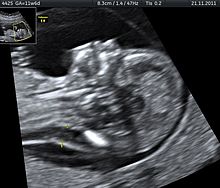
L'ecografia può essere utilizzata per lo screening della sindrome di Down. Reperti che possano essere indici di un aumento del rischio, quando l'esame viene effettuato tra la 14ª e la 24ª settimana di gestazione, sono: l'osso nasale piccolo o la sua mancanza, ventricolomegalia, spessore della plica nucale e un'arteria succlavia. La presenza o l'assenza di più segni offre una valutazione più precisa. L'aumento della translucenza nucale fetale indica un aumentato rischio di sindrome di Down nel 75-80% dei casi e di essere un falso positivo nel 6%.
Esami del sangue
Diversi valori del sangue possono essere misurati per prevedere il rischio di sindrome di Down durante il primo o il secondo trimestre. Solitamente si raccomanda di ripetere i test in entrambi i trimestri per poi raffrontarli con i risultati ecografici. Nel secondo trimestre spesso due o tre test vengono utilizzati in combinazione con due o tre test di: α-fetoproteina, estriolo non coniugato, hCG totale, e βhCG libero, riuscendo a rilevare circa il 60-70% dei casi.
L'esame del sangue della madre per il DNA fetale è in fase di studio e sembra essere promettente se eseguito nel primo trimestre. La "Società Internazionale per la Diagnosi Prenatale" ritiene giustificata la scelta dello screening per quelle donne le cui gravidanze sono ad alto rischio di riscontrare un caso di trisomia 21. È stata segnalata una precisione al 98,6% nel primo trimestre di gravidanza. Il test di conferma con tecniche invasive (amniocentesi, CVS) è ancora tuttavia necessario per confermare il risultato dello screening.
Diagnosi
Diagnosi pre-natale
Quando ci si trova in una situazione ad alto rischio di incorrere nella sindrome di Down, è necessario eseguire test diagnostici più invasivi, come l'amniocentesi, il prelievo dei villi coriali (CVS - Chorionic villus sampling) o il prelievo di sangue dal cordone ombelicale (PUBS - Percutaneous umbilical cord blood sampling), per confermare la diagnosi. L'efficacia dello screening è migliorata notevolmente verso la fine degli anni 1990: il tasso di rilevamento è tra il 90% e il 95%, mentre il rischio di incorrere in falsi positivi si attesta al 2% ÷ 5%. L'amniocentesi e il prelievo dei villi coriali sono i test più affidabili, ma comportano un aumentato di rischio di aborto spontaneo tra lo 0,5% e l'1%. Il rischio di incorrere in problemi agli arti aumenta nella prole a causa del test diagnostico: più precocemente viene eseguito, più il rischio aumenta, quindi l'amniocentesi non è raccomandata prima di 15 settimane di gestazione e il prelievo dei villi coriali prima delle 10 settimane.
Diagnosi post-natale
L'aspetto fisico del bambino appena nato può indurre dei sospetti sulla presenza della sindrome: l'esame clinico da parte di un pediatra può spesso confermare o smentire questi sospetti.
I criteri diagnostici comprendono 8 segni: faccia piatta, displasia dell'orecchio, protrusione della lingua, angoli della bocca rivolti verso il basso, ipotonia, eccesso di pelle del collo, piega epicantica e ampio divario tra alluce e secondo dito del piede. Se nel neonato sono presenti al massimo due di queste caratteristiche, si può stabilire che difficilmente ha la sindrome di Down (con meno di uno su 100 falsi negativi), se le caratteristiche presenti sono almeno 6 si può stabilire con ragionevole certezza che il neonato abbia la condizione (con meno di uno su 100.000 falsi positivi); nei casi intermedi invece la situazione non è chiara e sono allora consigliabili test genetici. Nei casi in cui non vi siano motivi clinici per fare la diagnosi, è stato suggerito che i genitori possano essere tenuti all'oscuro del sospetto iniziale di sindrome nel bambino.
Prevenzione
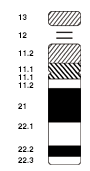
Quando si studiano casi di sindrome di Down dovuti a traslocazione, si trova di solito che un genitore, anche se fenotipicamente normale, presenta un cariotipo a 45 cromosomi. Uno di questi cromosomi è formato dai bracci lunghi del cromosoma 21, unito al braccio lungo di un altro cromosoma acrocentrico (di solito il cromosoma 14), mentre i bracci corti residui si uniscono a formare un piccolo cromosoma acentrico che solitamente è mitoticamente instabile e viene perduto, senza che vi siano conseguenze cliniche. Perciò ai genitori di un figlio affetto da sindrome di Down dovuta a sbilanciamento di traslocazione robertsoniana, si consiglia di procedere alla determinazione del loro cariotipo; se l'uno o l'altro genitore sono portatori della traslocazione, essi saranno avvisati del fatto che vi è un'aumentata probabilità di avere un altro figlio affetto da sindrome di Down.
Da diverso tempo si ritiene che la sindrome di Down, e ulteriori condizioni legate alla non-disgiunzione, sia più probabile in bambini nati da donne non giovani. Le ragioni di questo fatto non sono chiare ma si suppone che vi sia una correlazione con il tempo di permanenza della cellula uovo nell'ovaio, fermo nel diplotene della profase della meiosi 1, in cui si verifica lo scambio di frammenti di materiale genetico tra due cromatidi non fratelli di cromosomi omologhi. La lunga permanenza in questa fase di scambio determinerebbe la maggior probabilità di verificarsi errori nella disgiunzione dei cromosomi durante l'anafase.
Studi recenti hanno, tuttavia, mostrato che in circa il 20% di casi di sindrome di Down dovuta a non-disgiunzione, il cromosoma in più deriva dal padre (in metà dei casi durante la meiosi I e in metà durante la meiosi II) anziché dalla madre.
Un inadeguato apporto di acido folico nella madre sembra essere correlato con una maggior probabilità di avere un figlio con sindrome di Down. Per questo, alle donne incinte di età uguale o superiore ai 35 anni, viene consigliato di prendere dosi supplementari di questa sostanza.
Prognosi, prevenzione e monitoraggio delle complicanze

Fin dal 1981 la comunità medica ha formulato delle linee guida per il trattamento della sindrome, ma il programma più ampiamente accettato e diffuso è quello concepito dal Down Syndrome Medical Interest group (DSMIG). Questi programmi prevedono semplici strategie per la diagnosi precoce e una serie di protocolli per il monitoraggio della condizione e delle eventuali complicanze che possono insorgere, migliorando significativamente la prognosi di queste persone. Inoltre, i sempre più diffusi programmi di trattamento precoce e il progressivo mutamento che la società sta vivendo nei confronti della disabilità intellettiva, sono stati i motivi principali di una sempre maggior emancipazione da parte delle persone con trisomia 21. Grazie a questi cambiamenti, gli individui affetti dalla sindrome di Down hanno la possibilità di raggiungere una sufficiente autonomia, sia sul lavoro che nella vita sociale.
Molti bambini con sindrome di Down prendono il diploma di scuola superiore e sono in grado di svolgere un lavoro retribuito o di seguire una formazione universitaria. Le strategie di gestione, come l'intervento nella prima infanzia, lo screening per i problemi più comuni, cure mediche ove indicate, un ambiente familiare favorevole e una formazione professionale sono fattori in grado di migliorare lo sviluppo globale dei bambini con la condizione. Tuttavia crescere un bambino affetto dalla sindrome di Down potrebbe presentare maggiori difficoltà rispetto a crescerne uno privo della condizione.
Controlli raccomandati
Alcune organizzazioni sanitarie hanno formulato raccomandazioni per il controllo costante degli individui affetti da sindrome di Down, soprattutto per malattie particolari. Si raccomanda, inoltre, che esse vengano eseguite periodicamente.
Alla nascita, a tutti i neonati con la sindrome dovrebbe essere fatto un elettrocardiogramma e una ecografia del cuore. La riparazione chirurgica dei problemi cardiaci può essere richiesta fin dai primi tre mesi di età. Problemi legati alle valvole cardiache possono riscontrarsi nei giovani adulti e un'ulteriore valutazione ecografica può essere necessaria negli adolescenti e in coloro che hanno raggiunto l'età adulta. A causa dell'elevato rischio di cancro testicolare, alcuni medici consigliano il controllo dei testicoli annualmente.
| Test | Età pediatrica | Adulti |
|---|---|---|
| Udito | 6 mesi, 12 mesi, poi annualmente | 3–5 anni |
| T4 e TSH | 6 mesi e poi annualmente | |
| Vista | 6 mesi, poi annualmente | 3–5 anni |
| Denti | 2 anni, poi ogni 6 mesi. | |
| Malattia celiaca | Tra i 2-3 anni, o prima, se compaiono sintomi. | |
| Studio del sonno | Tra i 3-4 anni, o prima se compaiono sintomi della sindrome delle apnee ostruttive nel sonno. |
|
| Radiografia del collo | Tra i 3-5 anni |
La timpanostomia, un drenaggio che attraversa la membrana timpanica per eliminare l'essudato tipico dell'otite media acuta purulenta, si rende talvolta necessaria e spesso più di una volta durante l'infanzia per la probabilità di recidiva maggiore nei soggetti Down rispetto alla popolazione in generale. La tonsillectomia è spesso eseguita per evitare faringiti e apnee del sonno. La chirurgia non risolve sempre i casi di apnea del sonno, quindi è spesso necessario ricorrere ad una ventilazione meccanica a pressione positiva delle vie aeree (CPAP). La fisioterapia e la partecipazione alle attività di educazione fisica possono migliorare le abilità motorie. Tuttavia, non vi sono prove certe a sostegno di questa tesi, soprattutto negli adulti.
Devono essere fatti sforzi per evitare il virus respiratorio sinciziale umano tramite anticorpi monoclonali, in particolare per coloro che soffrono di problemi cardiaci. In coloro che sviluppano la demenza non vi è alcuna prova a favore, nonostante gli ampi studi effettuati, dell'utilizzo di memantina,donepezil,rivastigmina, o galantamina.
Sviluppo cognitivo
Gli individui con sindrome di Down differiscono notevolmente nella capacità di comunicazione tanto che lo screening di routine per i problemi dell'udito è altamente consigliato. L'uso di apparecchi acustici può essere di aiuto nei casi più gravi. Un intervento precoce sugli aspetti comunicativi può favorire le competenze linguistiche. Un ritardo nello sviluppo del linguaggio può richiedere un intervento logopedico per migliorare l'espressività.
Chirurgia plastica
La chirurgia plastica sui bambini con sindrome di Down è rara, e continua ad essere considerata controversa. La National Down Syndrome Society vede piuttosto come obiettivo quello di favorire il reciproco rispetto e l'accettazione, senza basarsi sull'aspetto esteriore delle persone con sindrome di Down.
Stato della ricerca
I meccanismi responsabili delle disabilità nelle persone affette da sindrome di Down sono al momento sconosciuti, nonostante gli studi condotti sul genoma umano, che iniziano a far chiarezza sui geni coinvolti nella condizione. Gli studi sono stati condotti comparando il fenotipo di un individuo con trisomia 21 con un soggetto con monosomia 21, una condizione opposta alla sindrome di Down, ottenendo così dei modelli con sovra o sotto dosaggio dell'espressione genica legata a quel cromosoma.
Inoltre si compiono continui sforzi al fine di sviluppare metodi finalizzati a migliorare la capacità cognitiva in coloro che sono affetti dalla sindrome. Uno di questi metodi deriva dalla possibilità di utilizzare cellule staminali. Altri metodi, in fase di studio al 2013, includono l'uso di antiossidanti, inibitori della gamma secretasi, agonisti adrenergici e memantina. La ricerca viene spesso effettuata su un modello animale, il topo Ts65Dn. Studi effettuati nel 2013 hanno dimostrato la possibilità di inattivare la terza copia del cromosoma 21, responsabile della sindrome di Down, utilizzando lo specifico gene a RNA denominato XIST (da X-inactivation gene), espresso nell'embrione solo dal cromosoma X destinato a essere silenziato. Tale gene induce modifiche all'eterocromatina, costituita da genoma non codificante e altri cambiamenti strutturali che portano questo cromosoma a mutarsi nella sua forma inattiva denominata corpo di Barr, evitando così che vi sia un'espressione duplicata di geni situati sui due cromosomi X.
Tramite un enzima, XIST è stato introdotto in una coltura di cellule staminali pluripotenti derivate da pazienti con sindrome di Down ottenendo il silenziamento dei geni contenuti nella copia soprannumeraria del cromosoma 21; si è riscontrato un effetto notevole sulla funzionalità delle cellule con la correzione degli anomali schemi di crescita e differenziazione tipici delle cellule di soggetti con sindrome di Down.
Società e cultura
Etica

Alcuni sostengono che non sia etico non offrire lo screening per la sindrome di Down e quando i risultati dei test sono pronti è anche considerato immorale non metterne a conoscenza la madre.
Alcuni ritengono ragionevole che i genitori scelgano di avere un bambino che possa avere un più alto benessere psicofisico. Spesso questa posizione è criticata da chi sostiene che il valore delle persone con disabilità equivale quello delle persone sane. Il movimento per i diritti della disabilità non ha preso una posizione chiara sullo screening, ma alcuni membri ritengono che i test e l'aborto siano discriminatori. Alcuni sono favorevoli all'aborto se il feto presenta forte disabilità mentre altri esprimono contrarietà anche a questa decisione. In una ricerca condotta su un gruppo di 40 madri statunitensi, che avevano già avuto un bambino con sindrome di Down, la metà di loro avrebbe deciso di eseguire uno screening se si fossero trovate nuovamente in gravidanza.
All'interno del cristianesimo, alcuni gruppi protestanti ritengono l'aborto accettabile se un feto presenta sindrome di Down, mentre gli ortodossi e i cattolici ritengono l'atto abortivo inaccettabile. Alcuni di coloro che sono contrari allo screening vedono tale pratica come una forma di "eugenetica". Non c'è accordo all'interno dell'Islam per quanto riguarda l'accettabilità dell'aborto in coloro che hanno una gravidanza con sindrome di Down, alcuni paesi islamici permettono l'aborto, mentre altri negano tale scelta.
Gli Emirati Arabi Uniti presentano tassi di incidenza della condizione molto superiori alla media globale. Ciò si spiega con l'elevata crescita demografica di quella società nella quale le donne, fortemente incentivate ad avere una prole numerosa, sono spinte a concepire anche in età avanzata. L'aborto è vietato negli Emirati Arabi, a meno che non vi sia un oggettivo e certificato pericolo di vita per la madre e quindi la disabilità non viene considerata come un giustificato motivo. Le istituzioni religiose emiratiane, infine, affermano che un bambino con una disabilità è una delle creazioni di Dio e quindi meritevole della vita.
Per valutare i dati relativi al ricorso allo screening bisogna anche tener conto, in alcune società e culture, della possibile stigmatizzazione sociale in cui potrebbero incorrere le donne in base alla decisione presa.
Tassi di aborto
Uno studio risalente al 2002, ha dimostrato che il 91-93% delle gravidanze nel Regno Unito e in Europa con una diagnosi di sindrome di Down venivano interrotte. I dati segnalano che dal 1989 al 2006 la percentuale di donne che scelgono di interrompere la gravidanza, dopo la diagnosi prenatale della sindrome di Down, è rimasta costante, circa il 92%.
Negli Stati Uniti alcune ricerche hanno evidenziato rispettivamente tassi di aborto del 95%, 98% e 87%.
Gruppi di sostegno
A partire dalla fine della seconda guerra mondiale si sono venuti a costituire diversi gruppi a sostegno degli individui con sindrome di Down. Queste organizzazioni promuovevano l'inserimento dei bambini con trisomia 21 all'interno del sistema scolastico, favorivano una maggior comprensione della condizione tra la popolazione generale e si occupavano di dare sostegno alle famiglie. Tra queste organizzazioni si possono citare: la Royal Society for Handicapped Children and Adults (MENCAP) fondata nel Regno Unito nel 1946 da Judy Fryd, la Kobato Kai sorta in Giappone nel 1964, la National Down Syndrome Congress statunitense nata nel 1973 grazie a Kathryn McGee e collaboratori, e la National Down Syndrome Society sempre statunitense e fondata nel 1979.
La prima "Giornata Mondiale della Sindrome di Down" si è tenuta il 21 marzo 2006, riconosciuta dall'Assemblea generale delle Nazioni Unite nel 2011. Il giorno e il mese (21/3) sono stati scelti per correlarli rispettivamente alla particolare numerazione cromosomica e al 21, che indica il cromosoma alterato.
Bibliografia
- (EN) Josef Warkany, Congenital Malformations, Chicago, Year Book, 1971, pp. 313–14, ISBN 0-8151-9098-0.
- (EN) Mary Caroline Richards, Toward wholeness: Rudolf Steiner education in America, University Press of New England, 1980, ISBN 978-0-8195-6062-9.
- (EN) Meira Weiss, Conditional love: parents' attitudes toward handicapped children, 1994, ISBN 978-0-89789-324-4.
- (EN) Janet Carr, Down's Syndrome: Children Growing Up, Cambridge, Cambridge University Press, 1995, ISBN 0-521-46933-3.
- (EN) Ward O'Conor, John Langdon Down, 1828–1896, Royal Society of Medicine Press, 1998, ISBN 1-85315-374-5.
- (EN) Stephen J. McPhee, Lawrence M. Tierney e Maxine A. Papadakis, Current medical diagnosis & treatment 1999, Norwalk, Appleton & Lange, 1999, ISBN 0-8385-1550-9.
- (ES) Jean Adolphe Rondal, Juan Perera e Lynn Nadel (a cura di), Síndrome de Down: revisión de los últimos conocimientos, Espasa Calpe, S.A., 2000, ISBN 978-84-239-8997-3.
- (ES) Charles J. Epstein, El futuro de la investigación biológica en el síndrome de Down, 2000, ISBN 84-239-8997-6.
- (EN) Gordon Grant, Peter Goward, Paul Ramcharan, Malcolm Richardson, Learning Disability: A Life Cycle Approach to Valuing People, McGraw-Hill International, 2005, pp. 43–44, ISBN 978-0-335-22556-9.
- (ES) Fundació Catalana Síndrome de Down, Síndrome de Down: aspectos médicos actuales, Elsevier, 2005, ISBN 978-84-458-1504-5.
- (EN) Erik Parens, Surgically Shaping Children: Technology, Ethics, and the Pursuit of Normality, Baltimore, Johns Hopkins University Press, 2006, ISBN 0-8018-8305-9.
- (EN) Cecil R. Reynolds e Elaine Fletcher-Janzen, Encyclopedia of special education a reference for the education of children, adolescents, and adults with disabilities and other exceptional individuals, 3ª edizione, New York, John Wiley & Sons, 2007, ISBN 978-0-470-17419-7.
- (EN) Liang Cheng e David Y. Zhang, Molecular genetic pathology, Totowa, N.J., Humana, 2008, ISBN 978-1-59745-405-6.
- (EN) Richard Urbano, Health Issues Among Persons With Down Syndrome, Academic Press, 2010, ISBN 978-0-12-374477-7.
- (EN) Lennard J. Davis, The Disability Studies Reader, Routledge, 2010, ISBN 978-0-415-87374-1.
- (EN) David Wright, Downs: The history of a disability, Oxford University Press, 2011, ISBN 978-0-19-956793-5.
Voci correlate
Altri progetti
-
 Wikizionario contiene il lemma di dizionario «mongoloidismo»
Wikizionario contiene il lemma di dizionario «mongoloidismo» -
 Wikimedia Commons contiene immagini o altri file sul mongoloidismo
Wikimedia Commons contiene immagini o altri file sul mongoloidismo
Collegamenti esterni
- Down, sìndrome di, su Treccani.it – Enciclopedie on line, Istituto dell'Enciclopedia Italiana.
- Gennaro Fiore, MONGOLISMO, in Enciclopedia Italiana, Istituto dell'Enciclopedia Italiana, 1934.
- mongolismo / Down, sìndrome di-, su sapere.it, De Agostini.
- (EN) Sindrome di Down, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.
- Associazione Italiana Persone Down, su aipd.it.
- (EN) Down's syndrome - Treatment, su nhs.uk.
- (DE) Grundstein für Therapie bei Down-Syndrom?, su wissenschaft.de.
- (ES) Fundacion iberoamericana Down 21, su down21.org. URL consultato il 27 maggio 2014 (archiviato dall'url originale il 30 maggio 2014).
| Controllo di autorità | Thesaurus BNCF 8050 · LCCN (EN) sh85039232 · GND (DE) 4012849-0 · BNF (FR) cb11940791r (data) · J9U (EN, HE) 987007560194605171 · NDL (EN, JA) 00567829 |
|---|