
| Fibrosi polmonare idiopatica | |
|---|---|
| Specialità | pneumologia |
| Classificazione e risorse esterne (EN) | |
| OMIM | 178500 |
| MeSH | D054990 |
| MedlinePlus | 000069 |
| eMedicine | 363273 e 301226 |
La fibrosi polmonare idiopatica (o IPF, idiopathic pulmonary fibrosis) è una malattia cronica, invalidante e con esito fatale caratterizzata da un progressivo declino della funzionalità polmonare. Il termine fibrosi polmonare significa cicatrizzazione del tessuto, ed è la causa del peggioramento della dispnea (mancanza di fiato). La fibrosi si associa solitamente a una prognosi infausta. Il termine "idiopatica" viene utilizzato poiché la causa della fibrosi polmonare è ancora sconosciuta.
La IPF solitamente si manifesta in soggetti adulti di età compresa tra 50 e 70 anni, in particolare nei soggetti con un pregresso di abitudine al fumo, e colpisce in misura maggiore gli uomini rispetto alle donne.
La IPF appartiene a un ampio gruppo di oltre 200 malattie polmonari, conosciute come malattie polmonari interstiziali (ILD), caratterizzate dal fatto che colpiscono l'interstizio polmonare. L'interstizio, il tessuto tra gli alveoli nel polmone, è la sede più colpita da ILD. Tali disturbi, tuttavia, spesso non colpiscono solamente l'interstizio, ma anche gli spazi aerei, le vie aeree periferiche e i vasi sanguigni. Il tessuto polmonare nei soggetti con IPF presenta le caratteristiche istopatologiche proprie della polmonite interstiziale usuale (UIP). La UIP pertanto è la controparte patologica della IPF.
Nel 2011 sono state pubblicate nuove linee guida per la diagnosi e la gestione della IPF. La diagnosi di IPF necessita dell'esclusione di altre cause conosciute di ILD e la presenza di specifiche caratteristiche radiologiche, identificate attraverso la tomografia computerizzata ad alta risoluzione (HRCT). In un contesto clinico appropriato è possibile effettuare la diagnosi di IPF attraverso il solo uso della HRCT, evitando la necessità di una biopsia polmonare chirurgica.
Classificazione
La fibrosi polmonare idiopatica (IPF) appartiene a un ampio gruppo di oltre 200 malattie polmonari, conosciute come malattie polmonari interstiziali (ILD), caratterizzate dal fatto che colpiscono l'interstizio, il tessuto tra gli alveoli nel polmone. La IPF è una manifestazione specifica della polmonite interstiziale idiopatica (IIP), che è a sua volta una forma di ILD, conosciuta anche come interstiziopatia polmonare.
La classificazione delle IIP pubblicata nel 2002 dall'American Thoracic Society/European Respiratory Society (ATS/ERS) è stata aggiornata nel 2013. La nuova classificazione prevede la suddivisione delle Polmoniti Interstiziali Idiopatiche (IIP) in tre categorie principali: IIP primaria, IIP rara e IIP non classificabile. Le IIP primarie sono raggruppate in:
- IP fibrosanti croniche (includono la IPF e la polmonite interstiziale non specifica [NSIP]),
- IP correlate al tabagismo (ad es. bronchiolite respiratoria associata a malattia polmonare interstiziale [RB-ILD] e polmonite interstiziale desquamativa [DIP])
- IP acute/subacute (ad es. polmonite criptogenetica in via di organizzazione [COP] e polmonite interstiziale acuta [AIP]).
La diagnosi di IIP richiede l'esclusione di cause note di ILD. Alcuni esempi di ILD da causa conosciuta sono la polmonite da ipersensibilità, la istiocitosi polmonare a cellule di Langerhans, l'asbestosi e la collagenopatia vascolare. Tali malattie, tuttavia, spesso non colpiscono solamente l'interstizio, ma anche gli spazi aerei, le vie aeree periferiche e i vasi sanguigni.
La figura sottostante mostra la nuova classificazione delle IIP.

Epidemiologia
Sebbene sia rara, la IPF è la forma più comune di IIP. La prevalenza della IPF è stata stimata tra 14 e 42,7 ogni 100.000 persone, sulla base di un'analisi statunitense di dati raccolti dalle assicurazioni sanitarie, con alcune differenze che si basano sulle definizioni dei casi utilizzati in questa analisi. La IPF è più frequente negli uomini che nelle donne e viene solitamente diagnosticata in soggetti di età superiore ai 50 anni. È difficile determinare l'incidenza della IPF dal momento che manca un'applicazione uniforme dei criteri diagnostici. Nei 28 Paesi dell'Unione Europea, una serie di fonti stima un'incidenza sulla popolazione di 4,6–7,4 persone ogni 100.000, valore che indica che ogni anno saranno diagnosticati 30.000–35.000 nuovi casi di IPF.
Un recente studio osservazionale, di coorte, retrospettivo, che ha incluso pazienti diagnosticati accidentalmente con ILD tra il 2003 e il 2009 presso l'Ospedale Universitario di Aarhus in Danimarca ha rivelato che l'incidenza di ILD era pari a 4,1 per 100.000 abitanti/anno. La IPF è risultata la patologia più frequentemente diagnosticata (28%), seguita da ILD correlata a patologie del tessuto connettivo (14%), polmonite da ipersensibilità (7%) e polmonite interstiziale non specifica (NSIP) (7%). L'incidenza di IPF era 1,3 per 100.000 abitanti/anno.
A causa della distribuzione disomogenea della malattia in Europa, è necessario mantenere un costante aggiornamento dei dati epidemiologici attraverso un registro europeo per ILD e IPF.
Cause/fattori di rischio di IPF
La fibrosi polmonare idiopatica o IPF è per sua stessa definizione idiopatica (che significa che non ha una causa nota), tuttavia alcuni fattori ambientali e l'esposizione a determinati agenti hanno dimostrato di aumentare il rischio di ammalarsi di IPF. Il fumo di sigaretta è il fattore di rischio di IPF più riconosciuto e accettato, che aumenta il rischio di ammalarsi di circa il doppio. Anche alcune esposizioni ambientali come l'esposizione a polveri di metallo, di legno, di carbone, di silice, di pietra ed esposizioni occupazionali legate all'agricoltura/allevamento hanno dimostrato di aumentare il rischio di IPF. Vi sono alcune evidenze che associano le infezioni virali alla fibrosi polmonare idiopatica e ad altre malattie polmonari fibrotiche.
Eziologia e patogenesi
Nonostante la IPF sia stata ampiamente studiata, le sue cause sono tuttora sconosciute. La fibrosi della IPF è stata messa in correlazione con il fumo di sigaretta, fattori ambientali (ad es. fattori occupazionali, quali l'esposizione a gas, fumo, agenti chimici o polveri), altre condizioni mediche, tra cui la malattia da reflusso gastroesofageo (MRGE), o a una predisposizione genetica (forma familiare). Non tutti questi fattori sono tuttavia presenti in tutti i soggetti affetti da IPF, e non forniscono pertanto una spiegazione soddisfacente di questa patologia.
Si ritiene che la IPF sia il risultato di un processo alterato di guarigione di una lesione, che comprende e comporta un deposito anomalo ed eccessivo di collagene (fibrosi) nell'interstizio polmonare con una infiammazione minima associata.
Si ipotizza che nella IPF si verifichi un danno iniziale o ripetuto alle cellule polmonari, dette cellule epiteliali alveolari (AEC), che occupano gran parte della superficie alveolare. Quando le AEC di tipo I sono danneggiate o distrutte, si ritiene che le AEC di tipo II proliferino per coprire la membrana basale esposta. In un normale processo di riparazione, le cellule epiteliali alveolari di tipo II muoiono e le restanti cellule si diffondono e subiscono un processo di differenziazione per divenire AEC di tipo I. In presenza di condizioni patologiche e del fattore di crescita trasformante beta (TGFβ), i fibroblasti si accumulano in queste aree lese e si differenziano in miofibroblasti che secernono collagene e altre proteine. In passato si pensava che l'infiammazione fosse il primo fattore a determinare la cicatrizzazione del tessuto polmonare. I risultati più recenti, tuttavia, mostrano che lo sviluppo di foci fibroblastici precede l'accumulo di cellule infiammatorie e, di conseguenza, il deposito di collagene.
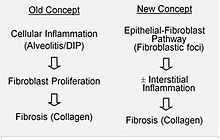
Tale modello patogenetico viene supportato indirettamente dalle caratteristiche cliniche della IPF, tra cui un'insorgenza insidiosa, la progressione per diversi anni, le esacerbazioni acute relativamente poco frequenti e la mancata risposta alla terapia immunosoppressiva. Attualmente diverse terapie che si concentrano sull'attivazione dei fibroblasti o sulla sintesi della matrice extracellulare, sono in fase di studio iniziale o se ne sta considerando lo sviluppo.
La IPF familiare rappresenta meno del 5% di tutti i casi di IPF e non è possibile distinguerla a livello clinico e istologico dalla IPF sporadica. Tra le associazioni genetiche vi sono le mutazioni delle proteine surfattanti del polmone A1, A2, C (SFTPA1, SFTPA2B) e della mucina (MUC5B). Una caratteristica importante della variante MUC5B è l'elevata frequenza della sua individuazione che si attesta al 20% circa nei soggetti del Nord e Ovest Europa e al 19% nella popolazione del Framingham Heart Study. Anche alcune mutazioni nei geni umani della telomerasi (trascrittasi inversa telomerasica o TERT, componente a RNA della telomerasi o TERC) vengono associate alla fibrosi polmonare familiare e, nel caso di alcuni pazienti, con la IPF sporadica. Recentemente è stata descritta in una famiglia con IPF una mutazione legata al cromosoma X in una delle subunità della telomerasi, dyskerin (DKC1).
Diagnosi
La diagnosi precoce di IPF è il prerequisito di un trattamento tempestivo che potenzialmente comporta un migliore risultato clinico a lungo termine di questa malattia progressiva e con esito fatale. In caso di sospetto di IPF, la diagnosi può essere difficile; si è dimostrato tuttavia che un approccio multidisciplinare che coinvolga pneumologi, radiologi e patologi esperti in malattie polmonari interstiziali contribuisca all'accuratezza della diagnosi di IPF. Un Documento di Consenso Multidisciplinare sulle polmoniti interstiziali idiopatiche è stato pubblicato dalla American Thoracic Society (ATS) e dalla European Respiratory Society (ERS) nel 2000, proponendo criteri specifici principali e minori per determinare la diagnosi di IPF. Nel 2011, tuttavia, sono stati pubblicati dei nuovi criteri semplificati e aggiornati per la diagnosi e la gestione della IPF da ATS ed ERS, in cooperazione con la Japanese Respiratory Society (JRS) e la Latin American Thoracic Association (ALAT). Attualmente i fattori che determinano una diagnosi di IPF sono:
- l'esclusione di cause conosciute di ILD, ad esempio l'esposizione ambientale domestica e professionale, disturbi del tessuto connettivo o esposizione/tossicità farmacologica;
- la presenza di un pattern radiologico tipico di UIP all'esame HRCT.
In un contesto clinico appropriato è possibile effettuare la diagnosi di IPF attraverso il solo uso della HRCT, evitando la necessità di una biopsia polmonare chirurgica.
Riconoscere l'IPF nella pratica clinica può essere difficoltoso dal momento che i sintomi che si presentano sono simili a quelli di patologie più comuni, quali l'asma, la broncopneumopatia cronica ostruttiva (BPCO) e lo scompenso cardiaco congestizio (www.diagnoseipf.com). La principale difficoltà per i medici è determinare se il pregresso, i sintomi (o segni), la radiologia e i test di funzionalità polmonare siano coerenti con una diagnosi di IPF o se invece i risultati siano determinati da un altro processo patologico. Per lungo tempo è stato difficile distinguere i pazienti con forme di ILD associate a esposizione all'amianto, a farmaci (quali gli agenti chemioterapici o la nitrofurantoina), ad artrite reumatoide e scleroderma/sclerosi sistemica dai pazienti affetti da IPF. Tra le altre considerazioni opportune per una diagnosi differenziale si annoverano le malattie polmonari interstiziali connesse a malattie del tessuto connettivo misto, sarcoidosi avanzata, polmonite da ipersensibilità cronica, istiocitosi polmonare a cellule di Langerhans e fibrosi da radiazioni.
Caratteristiche cliniche

In molti pazienti i sintomi si manifestano molto tempo prima della diagnosi. Tra le caratteristiche cliniche più comuni della IPF si elencano:
- età superiore a 50 anni;
- tosse secca e non produttiva sotto sforzo;
- dispnea da sforzo progressiva (difficoltà a respirare durante l'esercizio fisico);
- crepitii inspiratori bibasali secchi, che ricordano il suono del velcro, rilevati all'auscultazione (lo stetoscopio rileva un suono nei polmoni durante l'inalazione simile a una lenta apertura del velcro); ippocratismo digitale, una deformazione della punta delle dita di mani o piedi (vedi immagine);
risultati dei test di funzionalità polmonare non nella norma, con evidenza di restrizione bronchiale e difetto nella diffusione alveolare dei gas.
Tali caratteristiche sono dovute a una cronica carenza di ossigeno nel sangue e si possono verificare in numerose altre forme di malattie polmonari, senza essere specifiche della IPF. È tuttavia opportuno prendere in considerazione la presenza di IPF in tutti i pazienti con dispnea cronica da sforzo che manifestano tosse, crepitii inspiratori bibasilari o ippocratismo delle dita.
Valutare la presenza di crepitii simili al suono del velcro attraverso l'auscultazione dei polmoni è un metodo pratico per favorire una diagnosi precoce di IPF. I crepitii sottili sono facilmente riconosciuti dai medici e sono caratteristici della IPF.
Se sono presenti dei crepitii sottili bilaterali per l'intera durata dell'atto respiratorio e questi persistono dopo diversi respiri profondi, e in caso si manifestino nuovamente in diverse occasioni a diverse settimane di distanza in un soggetto di età ≥ 60 anni, è opportuno prendere in considerazione l'ipotesi di IPF e di conseguenza valutare l'esecuzione di un esame HRCT (TC ad alta risoluzione) del torace, più sensibile rispetto a una radiografia toracica. Dal momento che i crepitii non sono specifici della IPF, è necessario avviare un percorso diagnostico approfondito.
Radiologia
La radiografia toracica è utile per la routine di follow-up dei pazienti con IPF. La sola RX toracica purtroppo non è determinante ai fini diagnostici ma può tuttavia individuare una diminuzione del volume polmonare, solitamente con evidenti segni interstiziali reticolari alla base dei polmoni.

La valutazione radiologica attraverso HRCT è un punto focale nel percorso diagnostico della IPF. La HRCT viene effettuata utilizzando un comune tomografo assiale computerizzato senza iniettare mezzo di contrasto. Le sezioni dell'analisi sono molto sottili(1–2 mm).
La TC ad alta risoluzione (HRCT) del torace nei pazienti con IPF mostra tipicamente la presenza di alterazioni fibrotiche in entrambi i polmoni, con prevalenza alla base e nelle aree periferiche. Secondo le linee guida congiunte di ATS/ERS/JRS/ALAT del 2011, la HRCT è un elemento essenziale del percorso diagnostico della IPF in grado di individuare le forme di UIP attraverso la presenza di:
- opacità reticolari, spesso associate a bronchiectasie da trazione;
- polmone a nido d'ape (honeycombing) caratterizzato da raggruppamenti di spazi aerei cistici, con diametro solitamente simile (3–10 mm), occasionalmente più ampie. Generalmente a livello sub-pleurale e caratterizzato da pareti ben definite e con cisti disposte su almeno due file. Solitamente una sola fila di cisti non è sufficiente per definire l'aspetto a nido d'ape;
- opacità a vetro smerigliato, frequente ma meno estesa del pattern reticolare;
- caratteristica distribuzione alla base e periferica, sebbene spesso non uniforme.

Istologia
Secondo le linee guida aggiornate del 2011, in assenza di un pattern tipico di UIP all'esame HRCT, è necessario effettuare una biopsia polmonare per una diagnosi certa. I campioni istologici per la diagnosi di IPF devono essere prelevati almeno in tre diversi punti ed essere sufficientemente grandi al fine di consentire al patologo di valutare l'architettura polmonare sottostante. I frammenti bioptici di dimensioni ridotte, come quelli ottenuti con la biopsia polmonare transbronchiale (effettuata durante la broncoscopia) spesso non sono sufficienti a questo scopo. Pertanto, sono solitamente necessari campioni bioptici di dimensioni maggiori prelevati chirurgicamente attraverso una toracotomia o una toracoscopia.
Il tessuto polmonare nei soggetti con IPF presenta solitamente un pattern istopatologico caratteristico di UIP ed è pertanto la controparte patologica della IPF. Sebbene una diagnosi patologica di UIP spesso corrisponda a una diagnosi clinica di IPF, è possibile osservare un pattern istologico di UIP anche in altre patologie e fibrosi di origine conosciuta (ad esempio nelle patologie reumatiche). Esistono quattro caratteristiche chiave delle UIP che comprendono la fibrosi interstiziale in "conformazione irregolare", lesioni interstiziali, strutture a nido d'ape e foci fibroblastici.
I foci fibroblastici sono accumuli compatti di miofibroblasti e tessuto cicatriziale; assieme all'aspetto a nido d'ape sono i principali dati patologici che consentono la diagnosi di IPF.

Lavaggio broncoalveolare
Il lavaggio broncoalveolare (BAL) è una procedura diagnostica ben tollerata nell'ambito delle ILD. L'analisi citologica con BAL (conta cellulare differenziale) può essere presa in considerazione per la valutazione dei pazienti con IPF a discrezione del medico curante sulla base della disponibilità ed esperienza di tale procedura nella propria struttura. Il BAL può portare a diagnosi alternative specifiche: malignità, infezioni, polmonite eosinofila, istiocitosi X, o proteinosi alveolare. Nella valutazione dei pazienti con sospetta IPF, l'applicazione più importante del BAL risiede nell'esclusione di altre diagnosi. Un aumento isolato dei linfociti esclude la diagnosi di IPF.
Test di funzionalità polmonare
La spirometria individua tipicamente una riduzione della capacità vitale (VC) con una riduzione proporzionale della ventilazione o un aumento della ventilazione per la capacità vitale analizzata. Quest'ultimo dato è indicativo di una maggiore rigidità polmonare (minore adattamento polmonare) associata a fibrosi polmonare, che porta a una maggiore forza di retrazione elastica del polmone.
La misurazione del volume polmonare statico attraverso la pletismografia corporea o altre tecniche, rivela tipicamente una riduzione del volume polmonare (restrizione). Ciò è rappresentativo delle difficoltà incontrate nell'espansione di polmoni fibrotici.
La capacità di diffusione del monossido di carbonio (DLCO) è invariabilmente ridotta nella IPF e potrebbe essere l'unica anomalia nella patologia precoce o lieve. Tale insufficienza è alla base della propensione dei pazienti con IPF a manifestare una desaturazione di ossigeno durante l'esercizio fisico, che può essere analizzata anche tramite l'utilizzo del test del cammino di 6 minuti (6MWT).
Termini quali "lieve" o "moderata" e "grave" sono a volte utilizzati per classificare la malattia e si basano solitamente sulle misurazioni dei test di funzionalità polmonare a riposo. Non vi è tuttavia un chiaro consenso sulla classificazione dei pazienti affetti da IPF e su quali siano i migliori criteri e valori da applicare. La IPF da lieve a moderata è definita attraverso i seguenti criteri funzionali:
- capacità vitale forzata (FVC) ≥50%;
- DLCO ≥30%;
- distanza percorsa nel 6MWT ≥150 metri.
Programma di consulenza genetica per la IPF familiare
Si stima che il 10-15% dei pazienti affetti da IPF presenti una malattia di tipo familiare, ovvero ricorrente nell'albero genetico. Uno studio recente ha identificato le mutazioni genetiche associate alla fibrosi polmonare familiare (vedi sopra) che ora possono essere rilevate grazie ai test diagnostici sviluppati e resi disponibili al pubblico. La consulenza genetica fornisce informazioni sulla natura, il carattere ereditario e le implicazioni di una malattia genetica allo scopo di aiutare i singoli individui e le famiglie, nelle scelte sia personali sia mediche e di stabilire il loro rischio di sviluppare una malattia ereditata. Consulenza genetica e screening per la rilevazione di tutte le mutazioni conosciute dovrebbero essere effettuati soprattutto quando più di un membro della famiglia è affetto da fibrosi polmonare. Questo programma permette un'interpretazione personalizzata dei risultati e del loro impatto sia sulla salute del paziente sia sugli altri membri della famiglia.
Prognosi

Il decorso clinico della IPF può essere difficile da predire. La progressione della IPF è associata a un tempo medio di sopravvivenza stimato di 2-5 anni dopo la diagnosi.
La sopravvivenza a 5 anni alla IPF varia dal 20 al 40%, con un tasso di mortalità superiore a numerose neoplasie, tra cui il cancro al colon, il mieloma multiplo e il cancro della vescica.
Recentemente sono stati proposti un indice multidimensionale e un sistema di classificazione per predire la mortalità nella IPF. Il nome dell'indice è GAP (Gender, Age, Physiology), e si basa su genere, età e due variabili fisiologiche polmonari (FVC e DLCO) solitamente valutate nella pratica clinica al fine di predire la mortalità della IPF. Il valore maggiore dell'indice GAP (grado III) è risultato associato a un rischio di mortalità del 39% al primo anno.
Questo modello è stato anche applicato nella IPF e altre ILD, rivelandosi un buon fattore predittivo di mortalità in tutti i principali sottotipi di ILD. È stato sviluppato un indice ILD-GAP modificato, applicabile ai sottotipi di ILD, che fornisce una stima specifica della sopravvivenza. Nei pazienti con IPF, nonostante il tasso di mortalità globale a 5 anni sia elevato, il tasso annuale di mortalità per tutte le cause è relativamente basso in caso di compromissione della funzionalità polmonare da lieve a moderata. Per questo motivo, gli studi clinici della IPF a un anno valutano la variazione della funzionalità polmonare (FVC) piuttosto che la sopravvivenza. Oltre ai parametri clinici e fisiologici che predicono la velocità di progressione della IPF, le caratteristiche genetiche e molecolari sono correlate con la mortalità per la IPF. È stato dimostrato, infatti, che i pazienti che presentano uno specifico genotipo all'interno del polimorfismo del gene della mucina MUC5B mostrano un più lento declino della FVC e un miglioramento significativo della sopravvivenza. Nonostante l'importanza scientifica di questi dati, l'applicazione di un modello predittivo basato sulla valutazione di specifici genotipi non è a oggi possibile nella pratica clinica. Pirfenidone, una molecola a basso peso molecolare, è approvato non solo in Europa, ma anche in Giappone, Sud Corea, Canada, Cina, India, Argentina e Messico.
Trattamento
Gli obiettivi del trattamento della IPF sono essenzialmente la riduzione dei sintomi, l'arresto della progressione della malattia, la prevenzione delle esacerbazioni acute e il prolungamento della sopravvivenza. Il trattamento preventivo (ad esempio i vaccini) e i trattamenti sintomatici dovrebbero essere iniziati precocemente in ogni paziente.
Interventi farmacologici
Nel passato sono stati studiati numerosi trattamenti per l'IPF, tra cui l'interferone gamma-1β, il bosentan, l'ambrisentan, e gli anticoagulanti; tali trattamenti tuttavia oggi non sono più considerati delle opzioni terapeutiche valide. Molti di questi studi iniziali si basavano sull'ipotesi che la IPF fosse una patologia infiammatoria.
Pirfenidone
Il pirfenidone è una piccola molecola che combina sia effetti anti-infiammatori sia anti-ossidanti e anti-fibrotici in modelli sperimentali di fibrosi. Il pirfenidone, presente sul mercato con il nome commerciale Esbriet, è approvato in Europa per il trattamento dei pazienti con IPF da lieve a moderata. È approvato anche in Giappone e Corea del Sud (nome commerciale Pirespa), così come in Canada, Cina, India, Argentina e Messico.
Il pirfenidone è stato approvato nell'Unione Europea sulla base dei risultati di tre studi di fase III randomizzati, in doppio cieco, placebo controllati: uno condotto in Giappone, gli altri due in Europa e negli Stati Uniti (studi CAPACITY).
Una review sulla Cochrane Library (la rivista della Cochrane Collaboration for evidence-based Medicine) basata su quattro studi che hanno coinvolto 1155 pazienti in cui veniva confrontato il pirfenidone con il placebo, ha dimostrato una riduzione significativa del 30% nella progressione della malattia nei pazienti trattati con pirfenidone. Anche i valori di FVC o VC risultavano significativamente migliorati con pirfenidone, sebbene si sia dimostrato un lieve calo di FVC in solamente uno dei due studi CAPACITY. Sulla base di tali risultati contrastanti, la American Federal Food and Drug Administration (FDA) ha richiesto un terzo studio clinico di fase III, ASCEND, attualmente in corso negli Stati Uniti. Questo studio, completato nel 2014 e pubblicato online su New England Journal of Medicine, ha dimostrato che pirfenidone riduce in modo significativo il declino della funzionalità polmonare e la progressione della IPF. I risultati dello studio ASCEND sono stati, inoltre, aggregati a quelli dei due studi CAPACITY in un'analisi prespecificata che ha dimostrato che pirfenidone riduce la mortalità di circa il 50% nel corso di un anno di trattamento. Sulla base di questi risultati, pirfenidone ha ricevuto la definizione di Breakthrough Therapy dall'FDA, definizione riservata a quei farmaci destinati al trattamento di patologie gravi o mortali i cui dati clinici preliminari hanno dimostrato un sostanziale miglioramento di uno o più endpoint rispetto alle terapie esistenti.
L'azienda farmaceutica che ha sviluppato pirfenidone, InterMune Inc., ha inizialmente fornito il farmaco per uso compassionevole negli Stati Uniti e in Europa, già nel periodo precedente all'approvazione alla commercializzazione grazie a un programma di accesso allargato (Expanded Access Program, EAP). Dal 2014 il farmaco è regolarmente prescrivibile in Italia da parte di Centri specialistici individuati a livello delle singole regioni. Attualmente il farmaco è commercializzato dalla Roche dopo l'acquisizione della InterMune.
N-acetilcisteina e la tripla terapia
La N-acetilcisteina (NAC) è un precursore del glutatione, un antiossidante. Si ipotizza che il trattamento con dosi elevate di NAC possa agire sullo squilibrio ossidante-antiossidante che si verifica nel tessuto polmonare dei pazienti con IPF. Nel primo trial clinico su 180 pazienti (IFIGENIA), è stato dimostrato che la terapia con NAC riduce il declino di VC e DLCO in 12 mesi di follow-up se assunto in combinazione con il prednisone e l'azatioprina (tripla terapia).
Più recentemente, un ampio studio controllato randomizzato (PANTHER-IPF) è stato avviato dal National Institutes of Health (NIH) negli Stati Uniti al fine di valutare la tripla terapia e la monoterapia con NAC nei pazienti con IPF. Questo studio ha rilevato che la combinazione di prednisone, azatioprina e NAC aumenta il rischio di morte e il numero di ospedalizzazioni; il NIH ha quindi annunciato nel 2012 la chiusura anticipata del braccio in tripla terapia dello studio PANTHER-IPF. Lo studio ha concluso che “acetilcisteina rispetto a placebo, non ha offerto alcun beneficio significativo per quanto riguarda il mantenimento della FVC in pazienti con fibrosi polmonare idiopatica con compromissione della funzionalità polmonare da lieve a moderata".
Questo studio ha anche valutato il solo trattamento con N-acetilcisteina (NAC). I dati ottenuti, recentemente pubblicati su New England Journal of Medicine, hanno dimostrato che la monoterapia con NAC non comporta benefici significativi nei pazienti con IPF da lieve a moderata.
Nintedanib (precedentemente BIBF 1120)
Una molecola in fase di sviluppo ha completato due studi clinici di Fase III (INPULSIS-1 e INPULSIS-2). Nintedanib è un triplice inibitore sperimentale dell'angiochinasi che ha come bersaglio i recettori tirosin-chinasici coinvolti nella regolazione dell'angiogenesi e nella patogenesi della fibrosi e della IPF: il recettore del fattore di crescita dei fibroblasti (FGFR), il recettore del fattore di crescita derivato dalle piastrine (PDGFR) e il recettore del fattore di crescita dell'endotelio vascolare (VEGFR). In entrambi gli studi di Fase III, nintedanib ha significativamente ridotto il declino della funzionalità polmonare di circa il 50% in un anno di trattamento. Per quanto riguarda il raggiungimento degli endpoint secondari, solo nello studio INPULSIS-2 si è verificato un significativo aumento del tempo (ritardo) alla prima riacutizzazione grave nel gruppo in trattamento con nintedanib rispetto a placebo. Questo incremento non si è verificato nello studio INPULSIS-1. Nintedanib, come pirfenidone, ha ricevuto l'approvazione alla compilazione del file FDA come Priority Review.
Opzioni terapeutiche in corso di sperimentazione
Le molecole attualmente in sperimentazione per la IPF in studi clinici di Fase II includono gli anticorpi monoclonali simtuzumab, tralokimab, lebrikizumab, FG-3019 pamrevlumab e l'antagonista del recettore dell'acido lisofosfatidico BMS-986020. È in corso, inoltre, uno studio di Fase II per la molecola STX-100. Queste molecole hanno come bersaglio diversi fattori di crescita e citochine che rivestono un ruolo nella proliferazione, nell'attivazione, nella differenziazione o nell'alterazione della sopravvivenza dei fibroblasti.
È possibile reperire ulteriori informazioni alla pagina www.ClinicalTrials.gov, un registro e banca dati di risultati di studi clinici con finanziamento pubblico e privato a partecipazione umana svolti nel mondo.
Interventi non farmacologici
Trapianto di polmone
Il trapianto polmonare può essere utile nei pazienti candidabili da un punto di vista fisico a essere sottoposti a un intervento di tale portata. È stato dimostrato che nei pazienti affetti da IPF il trapianto di polmone riduca il rischio di decesso del 75% rispetto ai pazienti che restano in lista d'attesa. Dall'introduzione del punteggio di allocazione del polmone (LAS), che classifica i candidati al trapianto sulla base della probabilità di sopravvivenza, la IPF è divenuta l'indicazione più frequente per il trapianto di polmone negli Stati Uniti.
I pazienti sintomatici affetti da IPF con età inferiore ai 65 anni e con un indice di massa corporea (BMI) ≤26 kg/m2 dovrebbero essere candidati al trapianto polmonare; non esistono tuttavia dei dati chiari che indichino il momento preciso per il trapianto. Sebbene controversi, i dati più recenti indicano che il trapianto bilaterale polmonare è prevalente rispetto al trapianto polmonare singolo nei pazienti con IPF. Il tasso di sopravvivenza a cinque anni dal trapianto di polmone è stimato tra 50% e 56%.
Nelle linee guida sulla IPF del 2011, l'ossigenoterapia, od ossigenazione integrativa per uso domestico, è divenuta una raccomandazione per i pazienti con ipossiemia a riposo clinicamente significativa. Sebbene non sia stato dimostrato che l'ossigenoterapia migliori la sopravvivenza nei pazienti con IPF, alcuni dati indicano un miglioramento nella capacità di svolgere esercizio fisico.
Riabilitazione polmonare
La stanchezza e la perdita di massa muscolare sono problematiche frequenti e debilitanti per i pazienti con IPF. La riabilitazione polmonare può alleviare i sintomi manifesti di IPF e migliorare la funzionalità stabilizzando e/o invertendo le caratteristiche extrapolmonari della malattia. Il numero di studi pubblicati sul ruolo della riabilitazione polmonare nella fibrosi polmonare idiopatica è ridotto, tuttavia nella maggior parte di questi studi sono stati riscontrati miglioramenti significativi a breve termine sulla tolleranza all'esercizio, sulla qualità di vita e sulla dispnea da sforzo. I principali programmi di riabilitazione prevedono esercizio fisico, regolazione dell'alimentazione, terapia occupazionale, supporto informativo, e terapia psicosociale.
Nella fase avanzata della malattia, i pazienti con IPF tendono a interrompere l'attività fisica a causa dell'aumento di dispnea. Dove possibile, è opportuno scoraggiare l'interruzione dell'attività fisica.
Cure palliative
Le cure palliative si concentrano soprattutto sulla riduzione dei sintomi e sul miglioramento della condizione del paziente, più che sul trattamento della malattia. Vi può essere il trattamento di sintomi in fase di peggioramento con l'utilizzo di oppioidi cronici per dispnea acuta e tosse. Anche l'ossigenoterapia può essere utile come cura palliativa della dispnea nei pazienti ipossiemici.
Tra le cure palliative vi sono anche il supporto fisico ed emotivo e il sostegno psicosociale per i pazienti e i loro familiari. Con la progressione della malattia, i pazienti possono essere spaventati e andare incontro ad ansia e depressione: è pertanto consigliato prendere in considerazione un supporto psicologico. Uno studio recente su pazienti ambulatoriali con ILD, tra cui la IPF, ha mostrato come il grado di depressione, la funzionalità generale (sulla base dei risultati del test di cammino) e la funzionalità polmonare contribuiscano alla gravità della dispnea.
È possibile prendere in considerazione l'uso di morfina in casi specifici di dispnea particolarmente acuta. La morfina può ridurre la dispnea, l'ansia e la tosse senza andare a ridurre in modo significativo la saturazione di ossigeno.
Gestione e follow-up
Sono frequenti i casi di diagnosi errata di IPF, perlomeno fino a che i dati fisiologici e/o di imaging non indichino la presenza di una ILD, determinando un ritardo nell'avvio delle cure appropriate. Considerando che la IPF è una patologia con una sopravvivenza media di 3 anni dalla diagnosi, sarebbe opportuno indirizzare tempestivamente qualsiasi paziente con ILD manifesta o sospetta a un centro con competenze specifiche. Sulla base della complessità della diagnosi differenziale, è estremamente importante, ai fini di una diagnosi accurata, uno scambio multidisciplinare tra pneumologo, radiologo e patologo esperti nella diagnosi di ILD.
A seguito della diagnosi di IPF e di un'adeguata scelta terapeutica sulla base dei sintomi e dello stadio della malattia, è opportuno effettuare uno stretto follow-up del paziente. A causa del decorso estremamente variabile della malattia e dell'elevata incidenza di complicanze, quali il cancro al polmone (fino al 25% dei pazienti con IPF), è indispensabile effettuare delle analisi di routine ogni 3-6 mesi, che includano la spirometria (pletismografia corporea), il test della capacità di diffusione, il 6MWT, oltre alla valutazione della dispnea, della qualità della vita e della necessità di ossigeno.
Inoltre, la maggiore consapevolezza delle complicanze e delle condizioni cliniche concomitanti spesso associate alla IPF, richiede una valutazione di routine delle comorbidità, la maggior parte delle quali riflette semplicemente malattie concomitanti di invecchiamento, l'uso di farmaci e la loro interazione e gli effetti collaterali.
Esacerbazioni acute
Le esacerbazioni acute della IPF (AE-IPF) vengono definite come un peggioramento immotivato o lo sviluppo di dispnea nell'arco di 30 giorni con la presenza di nuovi infiltrati radiologici all'esame HRCT, spesso sovrapposti a un pattern di fondo compatibile con UIP. L'incidenza precoce delle AE-IPF varia dal 10% al 15% di tutti i pazienti. La prognosi di AE-IPF è infausta, con un tasso di mortalità compreso tra il 78% e il 96%. Vanno escluse altre cause di AE-IPF, quali l'embolia polmonare, lo scompenso cardiaco congestizio, lo pneumotorace o l'infezione. L'infezione polmonare deve essere esclusa tramite l'aspirato endotracheale o il BAL.
Molti pazienti che subiscono un peggioramento acuto necessitano di trattamenti intensivi, soprattutto se lo scompenso respiratorio si associa a instabilità emodinamica, comorbidità importanti o ipossiemia acuta. Tuttavia, il tasso di mortalità durante l'ospedalizzazione è elevato. La ventilazione meccanica va introdotta solamente dopo un'attenta valutazione della prognosi a lungo termine del paziente e, dove possibile, tenendo in considerazione le sue volontà. Le attuali linee guida, tuttavia, scoraggiano l'utilizzo della ventilazione meccanica nei pazienti con scompenso respiratorio conseguente a IPF.
In altre specie
La IPF è stata individuata in diverse razze di cani e di gatti; la sua manifestazione più tipica si riscontra nel West Highland White Terrier. Gli animali che ne sono affetti, condividono molte delle manifestazioni cliniche dell'uomo, tra cui una maggiore e progressiva intolleranza all'esercizio fisico, una frequenza respiratoria più elevata e una possibile disfunzione respiratoria.La prognosi solitamente è infausta.
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file sulla fibrosi polmonare idiopatica
Wikimedia Commons contiene immagini o altri file sulla fibrosi polmonare idiopatica
Collegamenti esterni
- Associazione Italiana Malattie Interstiziali o rare del Polmone, su aimip.org. URL consultato il 21 novembre 2007 (archiviato dall'url originale il 13 novembre 2007).
- Comunità Fibrosi Polmonare Idiopatica (IPF), su rareconnect.org.
- Associazione Morgagni per le Malattie Polmonari - AMMP O.D.V.
- Pulmonary fibrosis foundation, su pulmonaryfibrosis.org. URL consultato il 29 agosto 2013 (archiviato dall'url originale il 3 settembre 2013).
- Registro europeo sulla IPF, su pulmonary-fibrosis.net.
- ILD CARE FOUNDATION, su ildcare.nl.
- Pulmonary fibrosis foundation, su pulmonaryfibrosis.org. URL consultato il 29 agosto 2013 (archiviato dall'url originale il 3 settembre 2013).
- IPF - British Lung Foundation, su blf.org.uk.
- The European IPF Registry (eurIPFreg) has become Europe's leading database of longitudinal data from IPF patients, including control groups of patients with other lung diseases, su pulmonary-fibrosis.net.
- Coalition for Pulmonary Fibrosis, su coalitionforpf.org. URL consultato il 26 settembre 2014 (archiviato dall'url originale il 6 novembre 2014).
- ILD CARE FOUNDATION´s activity is focused to increase knowledge, support research, contribute to prevention and provide counselling for interstitial lung diseases, su ildcare.nl.
- www.diagnoseipf.com, su diagnoseipf.com. URL consultato il 26 settembre 2014 (archiviato dall'url originale il 4 settembre 2014).
- KnowIPFNow.com.
- inIPF, su inipf.com.
- IPFtoday.com.
- ipfcharter.org. URL consultato il 2 agosto 2020 (archiviato dall'url originale il 22 ottobre 2014).
- Federazione Italiana IPF e Malattie Rare Polmonari - FIMARP onlus