

| Tumori del sistema nervoso centrale |
|---|
|
Famiglie di tumori secondo la classificazione WHO del 2007.
|
La neurooncologia è una branca della medicina che si occupa dello studio, della diagnosi e della cura dei tumori del sistema nervoso.
In particolare, questa disciplina si interessa al trattamento neurologico medico chirurgico ed oncologico di pazienti affetti da neoplasie primarie o metastatiche del sistema nervoso centrale, di quello periferico e da qualunque altra affezione o complicanza relativa al sistema nervoso di origine neoplastica, ovvero dai trattamenti effettuati per curare tale tipo di malattie. La presente trattazione riguarda le principali famiglie di tumori del sistema nervoso centrale. Si rinvia ad altre voci per l'analisi dei tumori del sistema nervoso periferico. Ugualmente l'esposizione delle complicanze è affrontata all'interno della voce relativa al particolare tumore o famiglia di tumori.
Tumori del sistema nervoso centrale – Tumori primari
In letteratura i tumori del sistema nervoso centrale (SNC) hanno una prima grossolana suddivisione in primari (cioè originatisi nel SNC) e metastatici (con origine in un altro organo). Di questi ultimi, anche se hanno una incidenza pari a circa 10 volte i primi, si farà cenno alla fine, rimandandone ad altre specifiche voci la trattazione estesa.
Il termine tumori cerebrali viene assai spesso usato per indicare genericamente le neoplasie che si sviluppano nella scatola cranica, anche se in maniera impropria. Ad esempio, i meningiomi, che comprimono ma raramente invadono il cervello, vengono comunque considerati tumori cerebrali, così come i tumori delle ghiandole pituitaria e pineale, le quali non fanno però strettamente parte del cervello, ma risiedono nella teca. La denominazione più appropriata sarebbe quella di tumori intracranici. Nel seguito si userà normalmente l'aggettivo "cerebrale"; la dizione "intracranico" verrà usata meno spesso o laddove vi sia il rischio di ambiguità.
|
Tab.1 - Distribuzione dei tumori del SNC per sottotipo istologico
CBTRUS 1998-2002 (N=63.968) | |
| Istologia | Percentuale |
| Glioblastoma* | 20,3 |
| Astrocitoma* | 9,8 |
| Oligodendroglioma* | 3,7 |
| Ependimoma* | 2,3 |
| Tumori embrionali (compreso il medulloblastoma) | 1,7 |
| Tumori della guaina dei nervi periferici | 8,0 |
| Meningioma | 30,1 |
| Linfoma | 3,1 |
| Craniofaringioma | 0,7 |
| Tumori della ghiandola pituitaria | 6,3 |
| Altri | 14,0 |
| (*) Nel SNC i gliomi rappresentano il 40% circa di tutti i tumori e il 78% circa dei tumori maligni. | |
I tumori primari del sistema nervoso centrale (Tabella 1) comprendono un variegato insieme di entità patologiche, ciascuna con una sua distinta storia naturale. Per il fatto che le neoplasie della glia costituiscono da sole quasi il 40% di tali tumori, si può operare una prima distinzione tra tumori gliali (gliomi) e tumori non gliali.
I gliomi più comuni sono gli astrocitomi (che originano dalle cellule astrocitiche della glia), gli oligodendrogliomi (dalle cellule oligodendrogliali) e gli ependimomi (dalle cellule ependimali). La Tabella 2 riporta il peso di ciascun elemento della famiglia dei gliomi.
|
Tab. 2 - Distribuzione dei gliomi per sottotipo istologico CBTRUS 1998-2002 (N=25.539) | |
| Istologia | Percentuale |
| Glioblastoma* | 50,7 |
| Astrocitoma anaplastico* | 7,9 |
| Astrocitoma diffuso* | 1,7 |
| Astrocitoma pilocitico* | 5,7 |
| Altri astrocitomi* | 9,1 |
| Oligodendroglioma | 9,2 |
| Ependimoma | 5,6 |
| Altri | 10,1 |
| (*) Gli astrocitomi (compreso il glioblastoma) rappresentano il 75% circa di tutti i gliomi. | |
La morfologia e le caratteristiche genetiche di ciascun tipo di glioma verranno messe in evidenza nelle sezioni dedicate a ciascun particolare tipo di neoplasia. Vale comunque la pena di anticipare una suddivisione tra gliomi circoscritti e gliomi diffusi. Esempi del primo gruppo sono le seguenti varianti astrocitarie, abbastanza non comuni: astrocitoma pilocitico, xantoastrocitoma pleomorfo e astrocitoma subependimale a cellule giganti. La maggior parte dei gliomi presenta caratteristiche di alta diffusione nella materia bianca, cosa che rende praticamente impossibile un loro estirpamento per via chirurgica. Le neoplasie non gliali hanno istologia sia benigna (come ad esempio, solitamente, i meningiomi e gli adenomi pituitari), sia maligna, come i tumori neuroectodermici primitivi e i medulloblastomi, i linfomi primitivi del SNC, ed i piuttosto rari tumori delle cellule germinali.
Epidemiologia

I tumori maligni primari del sistema nervoso centrale sono relativamente rari, rappresentano infatti circa il 2% di tutte le neoplasie non benigne. Ciò nonostante i tumori cerebrali, sia ad istologia benigna che maligna, sono una fonte non comune di alta morbosità e mortalità, anche a causa delle peculiarità degli organi intracraniali. Negli Stati Uniti vengono diagnosticati annualmente circa 43 800 nuovi casi di tumori cerebrali, di cui 3 410 riguardano bambini ed adolescenti. Di tutti questi nuovi pazienti, circa 12 760 moriranno. L'incidenza dei tumori cerebrali è di 14,8 nuovi casi all'anno su 100 000 individui, di cui circa la metà istologicamente benigni. Questi ultimi, se non possono essere trattati chirurgicamente o mediante radioterapia, possono risultare mortali a causa della progressiva crescita nello spazio chiuso della scatola cranica. Le femmine presentano un'incidenza leggermente più alta dei maschi (15,1 rispetto a 14,3 nuovi casi all'anno su 100 000 individui), probabilmente a causa della più alta incidenza di meningiomi nelle donne. I tumori maligni del sistema nervoso centrale sono la causa principale di morte per tumori solidi nei bambini e la terza causa di morte per cancro negli adolescenti e giovani adulti, tra i 15 e 34 anni.
Nella Tabella 1, che elenca i principali tumori del SNC, si noti che il meningioma rappresenta il più comune tumore intracranico benigno e il glioblastoma il più comune a istologia maligna.
Eziologia

Una predisposizione genetica alle neoplasie del sistema nervoso centrale è relativamente poco comune, anche se alcuni gliomi possono apparire tra le complicanze di diverse malattie familiari.
In particolare, la mutazione di alcuni geni oncosoppressori caratterizza diverse sindromi ereditarie, che mostrano un'aumentata suscettibilità allo sviluppo di tumori cerebrali. La neurofibromatosi di tipo 1 (mutazione del gene NF1), la sindrome di Turcot (mutazione di APC), sindrome di Gorlin (mutazione di PTCH) e la sindrome di Li-Fraumeni (mutazione di TP53 ovvero CHEK2) vengono associate al più alto rischio di sviluppare tumori cerebrali.
Sono difficili da individuare i fattori ambientali collegati a tumori cerebrali primari. In alcuni studi, l'esposizione al cloruro di vinile è stata associata ad un'aumentata incidenza di gliomi di alto grado (sul concetto di gradazione vedi oltre).
Le radiazioni ionizzanti sono l'unica, rara causa, ben individuata, di tumore cerebrale primario. In particolare, il trattamento radioterapico di bambini con tinea capitis, ovvero di pazienti affetti da leucemia linfocitica acuta, craniofaringioma o linfoma non Hodgkin è risultato associato ad un maggiore rischio di glioma. Infine, si registra un aumentato rischio di linfoma cerebrale primitivo (ma non di altri tipi di tumore del sistema nervoso centrale) tra i pazienti di AIDS.
Clinica
Segni e sintomi
La sintomatologia della neoplasia cerebrale è causata da effetto massa, infiltrazione parenchimale e distruzione tissutale.
La cefalea, il sintomo più comune, è legata all'effetto massa e ne risulta colpito il 35% circa dei pazienti. Il presentarsi di forti mal di testa in un paziente che non ne ha mai sofferto è frequentemente caratteristico, specie se gli attacchi di cefalea (o emicrania che sia) sono più forti al mattino e sono associati a nausea, vomito e a deficit neurologici. In pazienti che già soffrivano di cefalea, il variare della fenomenologia di tale disturbo o un aumento di frequenza o intensità degli attacchi può essere un segno della presenza di massa intracranica. Crisi epilettiche si verificano in circa un terzo dei pazienti di glioma, specialmente nei casi di tumori di basso grado (v. oltre). Tuttavia tali situazioni possono associarsi a qualsiasi tumore del SNC. Deficit neurologici focali sono legati alla ubicazione del tumore. In una percentuale tra il 15 e 20% dei pazienti di glioma si registra pure un'alterazione dello stato mentale.
Diagnostica per immagini
La presenza di una neoplasia cerebrale può essere efficacemente rivelata attraverso la tomografia computerizzata (TC) e la risonanza magnetica nucleare (RM). La RM presenta una maggiore sensibilità rispetto alla TC nell'identificazione delle lesioni; tuttavia non è sempre di facile accesso per il paziente e presenta alcune controindicazioni: non può essere effettuata in portatori di pacemaker, di protesi incompatibili con il campo magnetico, di clips metalliche ecc. La TC resta la metodica di elezione nella rivelazione di calcificazioni interne alle lesioni o di erosioni ossee della teca o della base cranica. L'uso del mezzo di contrasto, iodato in caso di TC, paramagnetico in caso di RM (gadolinio), permette l'acquisizione di informazioni sulla vascolarizzazione e sull'integrità della barriera emato-encefalica, una migliore definizione del nodulo tumorale rispetto all'edema circostante e consente di avanzare ipotesi sul grado di malignità. L'esame radiologico permette inoltre di valutare gli effetti meccanici (e le conseguenti modificazioni dei rapporti delle strutture encefaliche) derivanti dalla presenza della massa “estranea”: idrocefalo ed ernie, i cui effetti possono anche risultare letali. Tale diagnostica, infine, in sede di preparazione dell'intervento chirurgico, permette di precisare la sede della lesione e la vicinanza o l'infiltrazione del tumore in zone del cervello vitali (aree cosiddette “eloquenti”). A questo scopo la RM risulta più efficiente della TC per il fatto che è in grado di fornire immagini tridimensionali.

Gli strumenti di diagnostica per immagini evidenziano il fenomeno di alterazione dal punto di vista radiologico del tessuto neoplastico rispetto al normale parenchima cerebrale (modificazioni della densità elettronica dei materiali in caso di TC e dell'intensità di segnale per la RM). Come la maggior parte dei tessuti patologici, anche i tumori sono caratterizzati da un maggiore accumulo di acqua intracellulare. Alla TC appaiono ipodensi, ovvero di densità inferiore al parenchima cerebrale, alla RM appaiono ipointensi nelle immagini T1-pesate ed iperintensi in quelle DP- e T2-pesate.

In una radiografia la zona di cervello sano non dovrebbe segnalare particolari luminescenze. È naturale, quindi, che si ponga attenzione alle porzioni di maggior segnale di contrasto.
Nel tumore, in genere, la maggior quota di contrast enhancement è dovuta alla particolare barriera emato-tumorale, che permette il passaggio di iodio (TC) e di gadolinio (RM) nello spazio interstiziale extravascolare intratumorale: aumenta così il segnale (densità o intensità) del tumore. Si faccia comunque attenzione al fatto che il contrast enhancement non delimita con certezza la neoplasia dall'edema perilesionale: in effetti, nei gliomi infiltranti maligni (quali, ad esempio, il glioblastoma e l'astrocitoma anaplastico), il reperto anatomo-patologico mostra tessuto neoplastico persino oltre l'edema vasogenico (quello cioè causato dalla distruzione della barriera emato-encefalica da parte del tumore), uno stato clinico che non è facilmente evidenziabile attraverso la diagnostica a immagini.
La TC cerebrale tipicamente evidenzia una massa che può (oppure no) essere resa più luminescente (enhancement) mediante il mezzo di contrasto. Alla TC i gliomi di basso grado (vedi oltre) appaiono di solito isodensi rispetto al normale parenchima e quindi possono non mostrare enhancement col mezzo di contrasto. Allo stesso modo, le lesioni nella fossa cranica posteriore risultano difficilmente identificabili alla TC. Di conseguenza, i risultati di tale tomografia da soli possono non sempre essere sufficienti ai fini diagnostici.
Il ricorso alla RM (di maggiore sensibilità rispetto alla TC) risulta imperativo nei casi dubbi per la conferma della presenza di una neoplasia cerebrale.
Nelle immagini RM T1-pesate, un tumore intracranico appare come una lesione massiva che può (oppure no) diventare più luminescente dopo l'utilizzo del mezzo di contrasto. Comunque, si ha sempre un'anomalia di segnale nelle immagini T2-pesate, ad indicare la presenza di una neoplasia o di edema vasogenico. Di solito, una maggior luminescenza (contrast enhancement) è indice di neoplasia con maggior grado di malignità. Un anello di enhancement è caratteristico del glioblastoma (vedi immagine), dove la porzione luminescente corrisponde alla parte vitale del tumore maligno e la regione più scura (ipointensa in T1) corrisponde alla zona di necrosi tissutale.
Sia la risonanza magnetica sia la tomografia computerizzata sono in grado di fornire anche informazioni fisiologiche sulle lesioni tumorali ma, ad oggi, la sensitività e la specificità di tali metodiche sono troppo basse per costituire un protocollo semeiotico definitivo. Una diagnosi certa necessita del ricorso alla biopsia o alla resezione chirurgica, con conseguente esame istologico del tessuto.
Stadiazione
La maggior parte dei tumori intracranici primari rimane localizzata nella scatola cranica, quindi non risultano necessarie le procedure sistemiche di stadiazione.
I tumori neuroectodermici primitivi, il medulloblastoma, i tumori a cellule germinali del SNC ed il linfoma primario del SNC, invece, si diffondono frequentemente, tramite lo spazio subaracnoideo, alle leptomeningi. Risulta necessaria, pertanto, una RM spinale a tutti i pazienti con tali diagnosi.
Tumori gliali (Gliomi)
Astrocitomi
Per la gradazione (di malignità) degli astrocitomi, nel tempo sono stati proposti in letteratura diversi sistemi (si veda al proposito la voce Gradazione dei tumori del sistema nervoso centrale). Dal 1993 il sistema di gradazione a 4 livelli proposto dalla World Health Organization (WHO), è risultato quello più largamente accettato e diffuso. Esso si basa su quattro caratteristiche istologiche: aumentata cellularità, presenza di mitosi, proliferazione endoteliale, necrosi. Secondo il sistema di classificazione WHO (vedi la voce Classificazione dei tumori del sistema nervoso centrale), gli astrocitomi di grado I, quale l'astrocitoma pilocitico, sono tipicamente a istologia benigna; gli astrocitomi di grado II (detti diffusi) presentano la sola caratteristica istologica di aumentata cellularità e sono neoplasie a basso grado di infiltrazione; gli astrocitomi di grado III (anaplastici) mostrano anche significativa presenza di mitosi; negli astrocitomi di grado IV (glioblastoma) si evidenziano pure proliferazione endoteliale e/o necrosi.
Astrocitomi di basso grado

I tumori circoscritti comprendono l'astrocitoma pilocitico (con la variante pilomixoide), l'astrocitoma subependimale a cellule giganti e lo xantoastrocitoma pleomorfo. Si tratta di neoplasie poco comuni, a istologia benigna e guaribili molto spesso con la sola chirurgia. Anche se l'escissione è incompleta (per vari motivi), il tumore può rimanere indolente ovvero può essere trattato con successo tramite radioterapia. Nei rari casi in cui il trattamento locale fallisce, si ricorre al trattamento sistemico della chemioterapia, la cui portata, però, non è unanimemente riscontrata in letteratura; sembra essersi riscontrato un certo beneficio, nel caso dei bambini, con la combinazione di carboplatino e vincristina.

Alla TC gli astrocitomi diffusi di grado II appaiono come lesioni a bassa attenuazione, ovvero isointense. Alla RM (la metodica di preferenza) l'agente di contrasto può non far risaltare l'immagine di queste neoplasie, ovvero la loro luminescenza può apparire sottile e debole.
Un enhancement focale intenso può indicare aree di aumentata anaplasia. Quando possibile, si suggerisce il ricorso alla biopsia, allo scopo di ottenere campioni della porzione apparsa luminescente: la prognosi, infatti, è tipicamente connessa alla parte maggiormente anaplastica del tumore.
Nella maggior parte dei casi, i pazienti con astrocitomi diffusi sono giovani adulti (terza e quarta decade di età) che tipicamente accusano attacchi epilettici. Condizioni per una prognosi favorevole includono la giovane età, la grandezza del tumore inferiore a 5 cm e, quando possibile, una resezione chirurgica estesa della neoplasia. La sopravvivenza media è di circa 5 anni. Le recidive tarde sono relativamente comuni, per cui tali pazienti devono essere seguiti per almeno 15 anni.
Nonostante il loro corso relativamente indolente, la maggior parte di questi astrocitomi evolve verso lesioni caratterizzate da maggiore anaplasia, che non sono normalmente guaribili con chirurgia e radioterapia. La terapia per i pazienti con astrocitomi diffusi di basso grado non presenta comunque unanimità di consenso in letteratura. Il ruolo della resezione “completa” è argomento di dibattito nei contesti specialistici. I risultati di alcuni studi evidenziano che la massima escissione del tumore fornisce i migliori risultati. In realtà i casi di resezione completa riguardano spesso pazienti con tumori piccoli ed unilaterali, che non coinvolgono strutture critiche del cervello. Un approccio pragmatico, tutto sommato accettabile per la generalità dei casi, risulta quello di una escissione quanto più possibile estesa del tessuto neoplastico, evitando di causare deficit neurologici significativi.

La radioterapia effettuata immediatamente dopo la diagnosi ha mostrato di estendere il tempo in cui il paziente è libero da malattia prima della ricorrenza, rispetto alla situazione in cui il ciclo di radioterapia è ritardato fino al momento della progressione. Allo stato attuale, tuttavia, non c'è unanime convincimento che la radioterapia subito dopo la diagnosi migliori la sopravvivenza globale (overall survival) del paziente.
Per i pazienti con pochi o nessun sintomo, o con attacchi epilettici che sono controllabili con farmaci anticonvulsivi, è accettabile ritardare la radioterapia fino a quando la crescita del tumore non porti ad una situazione difficile da gestire. La ragione della radioterapia ritardata risiede spesso nel desiderio di ridurre il rischio di danno neurologico indotto dalla radioterapia stessa. Questa motivazione, tuttavia, non appare comunque inoppugnabile.
Due test clinici prospettici “randomizzati” non hanno dato risultato positivo nel tentativo di mostrare un maggior beneficio nella somministrazione di radioterapia ad alte dosi, rispetto a radioterapia a dosi inferiori. Il trattamento comunemente usato consiste nella somministrazione totale tra i 45 e i 54 Gy, effettuata con frazioni singole tra 1,8 e 2,0 Gy.
Il ruolo della chemioterapia adiuvante, per i pazienti di astrocitomi di basso grado, è ancora oggetto di studio. I risultati preliminari di un test clinico (di fase 3) che confronta la radioterapia da sola con radioterapia seguita da chemioterapia con procarbazina, lomustina e vincristina (PCV) mostrano che l'abbinamento radio-chemioterapia fornisce un maggior periodo di “sopravvivenza libera da malattia” ma non una maggiore sopravvivenza totale. Data la tossicità associata al protocollo PCV, da più parti si suggerisce invece l'uso della temozolomide, sia come terapia iniziale, sia alla ripresa di malattia.
Astrocitoma anaplastico

I pazienti con astrocitoma anaplastico presentano di solito crisi epilettiche, deficit neurologici focali, cefalee, modifiche della personalità. L'età mediana alla diagnosi è di circa 45 anni. La risonanza rivela generalmente la presenza di una lesione massiva con aumento di segnale di contrasto (enhancement), anche se vi sono casi in cui tale enhancement non viene evidenziato. La diagnosi è stabilita con l'esame istologico del materiale relativo alla lesione, prelevato tramite biopsia o resezione chirurgica. La presenza di mitosi consente di distinguere l'astrocitoma anaplastico dagli astrocitomi di basso grado. Gli astrocitomi anaplastici hanno un'elevata propensione ad un peggioramento in direzione anaplastica, quindi è necessario che il materiale da esaminare sia sufficiente da permettere di distinguere il tumore da un vero e proprio glioblastoma. In particolare, una diagnosi istologica di astrocitoma anaplastico in un paziente che alla risonanza presenta il classico anello di enhancement del glioblastoma, fa capire che il materiale portato all'esame non è rappresentativo della lesione. (Vedi anche il caso esposto nelle immagini sopra: un glioblastoma insorgente in un astrocitoma di basso grado.)

Indici di prognosi peggiore includono l'età più avanzata, una scarsa condizione fisica, un danno neurologico significativo. In generale, l'esito terapeutico è migliore con una resezione chirurgica “completa”, ma non è chiaro se tale miglior risultato è da associare all'intervento in sé o allo scenario clinico complessivo che ha permesso una tale resezione.
Il trattamento standard prevede all'inizio la massima asportazione possibile, cercando di non aumentare l'eventuale deficit neurologico. La radioterapia è pure uno standard nel trattamento, in quanto ha mostrato di prolungare il periodo di sopravvivenza.
Controverso è il ruolo della chemioterapia. Alcuni test clinici di fase 3 mostrano che i pazienti possono trarre beneficio dal trattamento chemioterapico aggiunto alla radioterapia (rispetto al caso di sola radioterapia), mentre altri studi non confermano tale situazione. L'uso della sola carmustina o del regime PCV (procarbazina, lomustina e vincristina) è risultato associato ad una maggiore sopravvivenza in uno studio del 1999
e in una recente meta analisi. Quest'ultima rileva un aumento assoluto di circa il 6% nella sopravvivenza a 1 e 2 anni nei pazienti sottoposti a chemioterapia. E la sopravvivenza a due anni risulta maggiore con radioterapia più chemioterapia (37%) rispetto alla sola radioterapia (31%).
In contrasto con il lavoro precedente però, un grosso studio clinico “randomizzato” non ha verificato alcun beneficio aggiuntivo nell'accoppiata radioterapia più PCV rispetto alla sola radioterapia.

Neanche nel caso di uso della temozolomide (che si rivela utile nel trattamento delle recidive) sembra chiaramente stabilito un beneficio supplementare da parte della chemioterapia aggiunta alla radioterpia.
La mediana di sopravvivenza mostra un intervallo che va da 24 mesi a più di 36 mesi. L'ampiezza di questo campo di variazione riflette i criteri di selezione dei pazienti.
In caso di recidiva il ricorso alla chemioterapia non pone dubbi: sia i regimi basati sulla nitrosurea, sia la temozolomide hanno mostrato efficacia. In tal senso va l'approvazione di quest'ultimo farmaco da parte della Food and Drug Administration statunitense. La risposta alla temozolomide è del 35% per pazienti che non hanno ricevuto chemioterapia in precedenza e del 20% per pazienti che sono al successivo regime di chemioterapia (in particolare dopo nitrosurea).
Glioblastoma
I Glioblastomi sono i tumori della glia più comuni. Possono comparire ex novo o derivare da un astrocitoma diffuso o da un astrocitoma anaplastico.
Oligodendrogliomi
Le neoplasie a base oligodendrogliale sono relativamente poco comuni, in quanto riguardano meno del 5% circa di tutti i tumori cerebrali primari e non più del 10-15% circa dei gliomi
(Vedi anche le Tabelle 1 e 2). Ciononostante sono molto importanti per la unicità nella sensibilità alla chemioterapia.
Questi tumori si distinguono in lesioni “a basso grado” e anaplastiche. L'oligodendroglioma anaplastico risulta caratterizzato da alta cellularità, polimorfismo nucleare, mitosi frequenti, abbondante proliferazione endoteliale e necrosi.
Circa la metà degli oligodendrogliomi sono contraddistinti da perdita di eterozigosi dei cromosomi 1p e 19q, una caratteristica tipica (patognomica) per la diagnosi. (Recentemente è stato mostrato che tale perdita di eterozigosi è secondaria a traslocazione pericentromerica sbilanciata.)
La maggior parte degli oligodendrogliomi nasce come tumore di basso grado. I gliomi misti, quali l'oligoastrocitoma e l'oligoastrocitoma anaplastico, contengono componenti sia oligodendrogliali che astrocitiche.
Oligodendroglioma/Oligoastrocitoma di basso grado

La sopravvivenza mediana per i pazienti di oligodendroglioma “puro” è di circa 10 anni; quella dei pazienti di oligoastrocitoma è di circa 8 anni (quindi intermedia tra quella di un puro oligodendroglioma e quella di un puro astrocitoma). La delezione (o la traslocazione) della coppia 1p/19q nel tumore è associata ad una sopravvivenza maggiore.
L'età media alla diagnosi è di 35 anni. La sintomatologia tipica annovera crisi epilettiche, ma possono pure segnalarsi deficit neurologici focali, modifiche della personalità ovvero gli altri sintomi di pressione endocranica (cefalea, vomito, ecc.). Questi tumori non sono normalmente visibili alla TC, quindi la metodica di elezione per la diagnostica per immagini risulta la risonanza magnetica. Alla RM sono visibili come aumentata intensità di segnale nelle immagini T2-pesate. Alle immagini T1-pesate il segnale può risultare attenuato e il contrast enhancement captato solo occasionalmente. Può essere o no presente la mancanza di segnale da calcificazione.

Tali neoplasie sono più indolenti delle corrispondenti astrocitarie e, come per gli astrocitomi di basso grado, anche per questi tumori non c'è accordo nella letteratura per quanto riguarda il trattamento ottimale. È stata avanzata l'ipotesi che il beneficio di una escissione il più completa possibile sia rilevante, ma è da tener presente che spesso si tratta di casi di tumori di dimensioni non cospicue in zone non vitali del cervello. I primi risultati di un test clinico europeo non ha mostrato benefici, in termini di sopravvivenza, della radioterapia fatta immediatamente dopo l'intervento chirurgico rispetto a quella ritardata sino alla comparsa di una sintomatologia che non si riesca a controllare farmacologicamente, benché nei casi di radioterapia immediata è stato evidenziato un maggior lasso di tempo di assenza di sintomi prima della nuova progressione del tumore.

Due altri test clinici non hanno evidenziato benefici in una radioterapia a più alte dosi rispetto a una a dosi “intermedie”.
Dati provenienti da diversi studi indicano che una terapia iniziale con temozolomide o PCV può rimpicciolire un oligodendroglioma o un oligoastrocitoma in una percentuale che varia dal 31% al 61% dei casi. Ma è ancora da stabilire se questa “risposta” alla chemioterapia migliori la sopravvivenza globale del paziente ovvero aumenti solo la durata del periodo di scarsa sintomatologia prima della successiva fase di progressione recidiva verso situazioni più complesse da gestire.
Un test clinico progettato per confrontare la radioterapia da sola con la radioterapia seguita da PCV verificò un periodo medio di sopravvivenza senza malattia superiore nel gruppo col PCV, ma nessuna differenza sostanziale in termini di sopravvivenza globale tra i due gruppi. Alla ricorrenza ad entrambi i gruppi veniva somministrato il PCV, significando con ciò che si ottiene lo stesso risultato indipendentemente da quando si comincia la chemioterapia (prima o dopo la ricorrenza).
Anche con la temozolomide si ottiene una riduzione dell'azione tumorale, ed essendo meno tossica del PCV, viene da più parti preferito il suo uso.
Per riassumere, il trattamento iniziale (cioè non al momento della recidiva) prevede il controllo dei sintomi con solo anticonvulsivi, la radioterapia da sola, la chemioterapia da sola o la combinazione di radioterapia più chemioterapia. Alla recidiva, chirurgia, radioterapia e chemioterapia svolgono tutte un ruolo importante. Una seconda resezione (o una prima, se non era mai stata fatta) può ridurre i sintomi. Se la radioterapia non era stata fatta nel trattamento iniziale, è probabile che risulti efficace contro la recidiva. Una risposta alla temozolomide si registra in circa il 50% dei pazienti che presentino ricorrenza dopo la radioterapia.
Oligodendroglioma anaplastico e Oligoastrocitoma


Oligodendroglioma anaplastico presentano sintomi tipici risultanti da effetto massa e crisi epilettiche. Nonostante la loro chemiosensibilità, la sopravvivenza mediana è di soli 3-5 anni. Il trattamento prevede il massimo asportazione seguito da radioterapia. Per quanto riguarda la chemioterapia, si segnala che in due recenti studi clinici di fase III, i risultati della radioterapia sono stati confrontati con quelli della radioterapia combinata e procarbazina, lomustina , vincristina chemioterapia. Sebbene il sopravvivenza senza sintomo rilevanti fosse più lungo con la terapia combinata, il sopravvivenza globale era lo stesso con entrambe le terapie. I pazienti con 1p/19q-Delezione hanno ottenuto i migliori risultati terapeutici, i pazienti senza 1p/19q deletion sono stati in grado di migliorare i loro risultati con la chemioterapia PCV.
Prospetticoe studio clinicon hanno dimostrato che circa il 50-70% dei pazienti con ricorrente oligodendroglioma anaplastico dopo radioterapia risponde positivamente a radioterapia chemioterapia con PCV o temozolomide. Sebbene non sia stata stabilita un'efficacia superiore della terapia con temozolomide e PCV, la mancanza di cumulativo e Mielosoppressione con temozolomide da utilizzare all'inizio del trattamento della ricaduta.
Ependimoma

L'ependimoma è una neoplasia che si sviluppa dalle cellule ependimali, che rivestono i ventricoli, il plesso corioideo, il filum terminale e il canale centrale del midollo spinale. Cellule ependimali sono pure presenti nel parenchima cerebrale quale risultato di migrazione da aree periventricolari alla corteccia cerebrale, durante lo stadio embrionale.
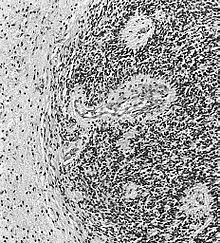
Questo tipo di tumore può comparire ad ogni età, ma presenta due picchi caratteristici, uno da zero a 10 anni ed un altro tra i 40 e i 50 anni. Le lesioni intracraniche (di solito nella fossa cranica posteriore) sono più comuni nella prima fascia di età, quelle spinali nella seconda.
Come si vede dalle Tabelle 1 e 2, si tratta di tumori abbastanza rari, sia in assoluto tra le neoplasie del sistema nervoso (2,3%), sia tra i gliomi (5,6%).
Si distinguono in lesioni di basso grado (I e II della scala WHO) e lesioni anaplastiche (III della scala WHO). In particolare il subependimoma e l'ependimoma mixopapillare sono di grado I,
l'ependimoma è di grado II, l'ependimoma anaplastico è di grado III. (Vedi la Classificazione dei tumori del sistema nervoso centrale.)
Gli ependimomi di basso grado nella spina dorsale resecabili vengono trattati con la sola chirurgia.
Mentre il ruolo della radioterapia postchirurgica per gli ependimomi intracranici di basso grado rimane controversa, i tumori anaplastici o quelli di basso grado non completamente escissi sono normalmente trattati con radioterapia.
Studi clinici hanno mostrato che gli ependimomi rispondono ai regimi chemioterapici, soprattutto a quelli basati sul platino. Dallo studio appena citato si evince infatti che la chemioterapia basata su platino fornisce il 67% delle risposte, mentre i regimi basati su nitrosurea hanno una risposta del 25%.
Per quanto riguarda la prognosi, gli ependimomi di grado II hanno una sopravvivenza libera da malattia a 6 anni del 66% ed una sopravvivenza globale dell'87%; per gli ependimomi anaplastici questi valori scendono rispettivamente al 29% e al 37%.
Tumori non gliali
Medulloblastoma
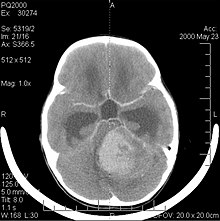
Il medulloblastoma, come altre neoplasie embrionali quali il tumore cerebrale neuroectodermico primitivo o il neuroblastoma cerebrale, è un tumore maligno del sistema nervoso centrale molto raro nella popolazione adulta (oltre 21 anni). È il tumore cerebrale maligno più frequente nell'infanzia, anche i giovani ne sono però a rischio. Il picco d'incidenza si verifica nei bambini di età tra i 2 e i 7 anni. Questo tumore è tipico della fossa cranica posteriore, ove si localizza in entrambi gli emisferi del cervelletto ovvero nel verme cerebellare ed essendo invasivo ed a rapida crescita usualmente diffonde ad altre parti del sistema nervoso centrale attraverso il liquor: può infiltrare il pavimento del vicino quarto ventricolo ed estendersi nella sua cavità, può anche passare nelle meningi. Più raramente, può dare metastasi extracraniche. La sintomatologia al presentarsi della neoplasia includono perdita di equilibrio, mancanza di coordinazione, diplopia, disartria e, a causa del coinvolgimento del quarto ventricolo (per il quale è comune un idrocefalo ostruttivo), i segni dell'idrocefalo, includenti cefalea, nausea, vomito, andatura instabile.
La risonanza magnetica usualmente rivela una lesione massiva a significativo contrast enhancement coinvolgente il cervelletto. Come sopra si diceva, il medulloblastoma ha un'alta propensione ad infiltrare focalmente le leptomeningi, così come a propagare attraverso lo spazio subaracnoideo per coinvolgere i ventricoli, la convessità cerebrale, le superfici leptomeningee spinali. Di conseguenza risulta necessario sottoporre a risonanza l'intero asse cranio-spinale.

È affidato alla chirurgia il compito di rimuovere quanto più è possibile della massa rappresentata dalla lesione, infatti residui tumorali postchirurgici sono causa di una prognosi peggiore. Foriera di prognosi non favorevole è anche la presenza di cellule neoplastiche nel liquido cerebrospinale ovvero la rilevazione alla risonanza di metastasi leptomeningee.
La chirurgia da sola di solito non è sufficiente come terapia, tuttavia lo può risultare in certi casi la successiva radioterapia all'asse cranio-spinale, con focalizzazione sul sito del tumore primario.
La chemioterapia dopo la radioterapia, inoltre, aumenta il tasso di guarigione. Si usano farmaci a base di platino (cisplatino o carboplatino), l'etoposide, e un agente alchilante (ciclofosfamide o lomustina) insieme alla vincristina.
Con un appropriato trattamento i casi di lunga sopravvivenza (superiore a 3 anni), per i pazienti di medulloblastoma, vanno dal 60% all'80%.
Meningioma

I meningiomi sono i tumori intracranici più diffusi (Vedi Tabella 1). Sono solitamente benigni ed originano dall'aracnoide, membrana che ricopre il cervello e il midollo spinale. L'incidenza di questo tipo di neoplasie è di circa 2 casi all'anno ogni 100.000 abitanti. Sono più comuni nelle donne, nella sesta e settima decade di vita. La loro frequenza è maggiore per i pazienti con neurofibromatosi di tipo 2.
La perdita del cromosoma 22 è caratteristica dei meningiomi, benché ancora non sia chiaro il significato prognostico di questa scoperta.
Nonostante che questa lesione abbia espressi recettori per androgeni, estrogeni, progesterone e somatostatina, le terapie dirette all'utilizzo di questi recettori non hanno ancora mostrato efficacia.
I pazienti con meningioma possono presentare la sintomatologia tipica di una lesione massiva nella scatola cranica, incluse crisi epilettiche e deficit neurologici focali.

Il meningioma, che può essere anche asintomatico, risulta talvolta scoperto incidentalmente da TC o risonanza magnetica, effettuate per altre ragioni. Questo tumore alla risonanza ha un aspetto caratteristico, che consiste, di norma, in un contrast enhancement uniforme lungo la dura, con netta separazione dal parenchima cerebrale. Altra caratteristica (benché non presente in tutti i casi) è la cosiddetta “coda durale”, rappresentato da enhancement che si estende oltre la lesione, ad indicare il punto di ancoraggio nella dura.
Solita è la presenza di edema peritumorale, conseguenza del fattore di crescita vascolare endoteliale secreto dalle cellule neoplastiche, che influenza a sua volta l'effetto massa locale.
Molti meningiomi scoperti incidentalmente non necessitano di trattamento al momento della diagnosi iniziale. Per i pazienti con mengiomi asintomatici può risultare appropriato e sufficiente tenere la lesione sotto osservazione. L'evidenza epidemiologica suggerisce che i due terzi di questi pazienti non avrà sintomatologia straordinaria.
Se si riscontra nel paziente un significativo effetto massa, che vi siano o no dei sintomi, il trattamento di elezione è normalmente la resezione completa. L'escissione è spesso realizzabile se il meningioma è situato sulla convessità cerebrale, il solco olfattivo, il seno sagittale superiore o la fossa posteriore. La resezione può risultare molto più difficoltosa se il tumore si presenta in altri siti, quali le regioni sfenoidale, parasagittale, orbitale, tentoriale o del clivus. In tali circostanze per il controllo del tumore risultano oltremodo utili la radioterapia classica o la radiochirurgia stereotassica.
In uno studio della Mayo Clinic, che confrontò la percentuale di controllo del tumore dopo resezione chirurgica e con radiochirurgia, in pazienti con meningioma intracranico di dimensioni medio-piccole e senza sintomi da effetto massa,
la radiochirurgia risultò ottenere un migliore controllo del tumore (98% contro 88%) e con minori complicanze (10% contro 22%) rispetto alla escissione chirurgica.
La chirurgia stereotassica è normalmente riservata alle lesioni più piccole (cioè inferiori a 3–4 cm), laddove per lesioni più grandi ovvero prossime a strutture critiche, come i nervi ottici, si usa la radioterapia frazionata.
Per quanto riguarda la chemioterapia, ad oggi nessun intervento farmacologico ha mostrato efficacia antitumorale riproducibile.
Raramente i meningiomi presentano caratteristiche istologiche atipiche o di franca malignità. In questi casi però risultano altamente aggressivi. L'approccio a tali tumori è identico a quello visto per i tumori benigni, con la differenza che la radioterapia postchirurgica diventa usuale e non episodica.
Linfoma primario del SNC

I linfomi primari del sistema nervoso centrale (cioè che sorgono nel SNC in assenza di linfoma al di fuori del SNC al momento della diagnosi)
costituiscono approssimativamente dal 2% al 3% di tutti i tumori cerebrali dei pazienti con un normale sistema immunitario (Cfr. Tabella 1).
La neoplasia è più comune nei maschi dai 55 ai 60 anni; quasi metà di tutti i linfomi si hanno in pazienti che hanno più di 60 anni e circa un quarto in pazienti con più di 70. L'incidenza sembra che stia aumentando, anche se non è chiaro se tale aumento sia reale o rifletta un'alterazione di rilevazione.
Esposti a un maggior rischio di linfoma del SNC sono sicuramente i pazienti con un sistema immunitario compromesso, quindi coloro che hanno subito un trapianto d'organi, quelli che hanno un'immunodeficienza congenita o una malattia autoimmune o che sono infetti dal virus dell'AIDS. I linfomi cerebrali associati al virus dell'immunodeficienza sono collegati con il virus di Epstein-Barr, in particolare nei pazienti con un conteggio di linfociti CD4
inferiore a 500 cellule per millimetro cubo (di sangue).
La maggior parte dei linfomi del SNC sono del tipo a grandi cellule B.
I pazienti presentano una varietà di sintomi caratteristici di lesione massiva focale o multifocale. La risonanza mostra di solito tumori, con contrast enhancement omogeneo, all'interno della materia bianca periventricolare profonda. Multifocalità ed enhancement disomogeneo sono tipici in pazienti con sistema immunitario compromesso.
Estremamente importante è l'analisi del linfoma del SNC nella diagnosi differenziale delle neoplasie cerebrali. Si tenga conto che la somministrazione di corticosteroidi può dare come risultato la completa scomparsa dell'enhancement lesionale, rendendo difficoltosa la diagnosi. Di conseguenza, quando si consideri un linfoma del SNC in diagnosi differenziale occorre evitare i corticosteroidi, a meno che l'effetto massa non stia causando un serio ed immediato problema al paziente.
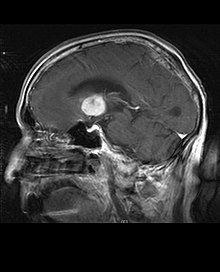
Criticamente importante è l'ottenimento di un campione bioptico della sospetta lesione, in quanto molte malattie del SNC, maligne e non, possono apparire un linfoma.
Diversamente dai linfomi sistemici “a cellule B grandi”, per i quali sia la chemioterapia che la radioterapia sono efficaci e il trattamento di lesioni localizzate è curativo, il linfoma del sistema nervoso centrale tipicamente risponde alla terapia iniziale ma poi recidiva. Come per il linfoma sistemico, il ruolo della chirurgia è ristretto soprattutto all'ottenimento di appropriati campioni di tessuto per la diagnosi.
La radioterapia dell'intero cervello (panencefalica) era una volta la strada maestra del trattamento. Sfortunatamente, anche con lesioni localizzate, la mediana di sopravvivenza con la sola radioterapia è di circa 1 anno. La recidiva interessa di solito il sito della precedente lesione oltre ad altre regioni. Interessanti sono le risposte con la chemioterapia.
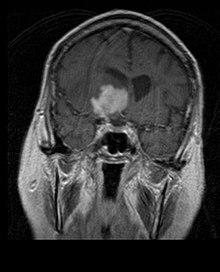
Hanno mostrato di fornire una migliore sopravvivenza globale, rispetto alla sola radioterapia, studi clinici nei quali è stato usato del metotrexato ad alte dosi, da solo, come primo trattamento e rinviando la radioterapia al momento della recidiva/progressione; il mix metotrexato, vincristina, procarbazina, metotrexato intratecale, radioterapia panencefalica, citarabina; ovvero la chemioterapia intraarteriosa (metotrexato per via intraarteriosa, ciclofosfamide ed etoposide per via intravenosa), dopo la modifica della barriera emato-encefalica con mannitolo.
Nei regimi con metotrexato la mediana di sopravvivenza è risultata di molto superiore a quella associata alla radioterapia da sola (intervallo da 24 a 40 mesi). In alcuni casi la radioterapia è usata solo alla recidiva, in caso di regressione iniziale ottenuta con la chemioterapia; sono riportati casi di lunga sopravvivenza anche senza l'uso di radioterapia.
Per la natura diffusa del linfoma del SNC, normale è l'affidamento alla radioterapia panencefalica, la quale però porta con sé un alto rischio di demenza, secondaria a leucoencefalopatia. Questo rischio potrebbe essere ridotto con lo sviluppo di strategie di controllo efficace del tumore che evitino la radioterapia panencefalica.
La terapia iniziale per i pazienti con sistema immunitario compromesso è la riduzione delle cause di immunosoppressione. La prognosi per questi pazienti è normalmente peggiore di quella per i pazienti con alla diagnosi un sistema immunitario normale.
A causa di infezioni concomitanti il tumore e una condizione fisica di solito non ottimale, in questi pazienti immunodepressi spesso la chemioterapia non può essere somministrata.
Come per le altre neoplasie cerebrali, la risposta ai trattamenti è correlata all'età ed alla condizione fisica.
Tumori del sistema nervoso centrale – Tumori metastatici
Metastasi cerebrali

Le metastasi al cervello sono le neoplasie intracraniche più comuni negli adulti 10 volte più frequenti dei precedenti tumori cerebrali primari.
Si verificano nel 20-40% degli adulti con cancro e sono associate soprattutto con il carcinoma polmonare e mammario e col melanoma.
Queste lesioni sono il risultato della propagazione di cellule cancerose attraverso il flusso sanguigno e sono massimamente presenti alla connessione della materia grigia con quella bianca, dove il calibro dei vasi sanguigni cambia, intrappolando così gli emboli cancerosi.
L'80% delle lesioni si verificano negli emisferi cerebrali, il 15% nel cervelletto e il 5% nel tronco encefalico.
Approssimativamente l'80% dei pazienti hanno una storia di cancro sistemico e il 70% presentano metastasi cerebrali multiple.
Sostanziali passi avanti si sono fatti negli ultimi tempi nella diagnosi e nel trattamento di queste lesioni, migliorando sopravvivenza e controllo della sintomatologia.
Segni e sintomi alla presentazione sono simili a quelli delle altre lesioni massive nel cervello.
Lo strumento diagnostico di elezione è la risonanza con mezzo di contrasto. Tuttavia, nei pazienti di cancro, non tutte le lesioni cerebrali sono metastasi. In uno studio clinico prospettico su pazienti con cancro sistemico sospetti di avere una metastasi cerebrale singola, l'11% dei campioni di tessuto mostrò invece un tumore cerebrale primario o un'infiammazione ovvero un'infezione.

Due studi prospettici randomizzati hanno mostrato che chirurgia più radioterapia panencefalica producono migliori risultati della sola chirurgia, in pazienti selezionati. Cioè in buone condizioni fisiche, con una lesione sistemica stabile o limitata e con una metastasi cerebrale singola chirurgicamente accessibile. Chirurgia più radioterapia danno come risultato un numero inferiore di decessi per cause neurologiche rispetto alla chirurgia da sola. Tuttavia l'aggiunta di radioterapia panencefalica non migliora la sopravvivenza globale rispetto alla sola chirurgia.

Per le lesioni difficilmente trattabili chirurgicamente può risultare efficace la radiochirurgia stereotassica. Due studi prospettici randomizzati hanno verificato che pazienti selezionati con un numero limitato di metastasi cerebrali hanno avuto maggior giovamento quando trattati con radiochirurgia più radioterapia panencefalica che con trattamento di sola radioterapia a tutto il cervello.
Un test clinico prospettico ha raffrontato l'efficacia di chirurgia e radiochirurgia, randomizzando i pazienti con singola piccola metastasi in un primo gruppo con chirurgia seguita da radioterapia panencefalica e un secondo gruppo con sola radiochirurgia. Non è stata trovata differenza significativa nei risultati. Analisi retrospettive hanno riportato risultati in conflitto.
Ricapitolando, la letteratura mostra risultati equivalenti per chirurgia e radiochirurgia. Quest'ultima sembra più conveniente, efficace e sicura per lesioni piccole o in regioni inaccessibili alla chirurgia. La radiochirurgia offre un'alternativa ragionevole a pazienti che non sono candidabili alla chirurgia per ragioni mediche. Tuttavia la chirurgia è chiaramente la modalità ottimale per ottenere tessuto per la diagnosi e per l'escissione di lesioni che causano effetto massa. Quindi, radiochirurgia e chirurgia andrebbero meglio considerate come due metodiche complementari ma differenti, da usare ciascuna a seconda della diversa situazione del paziente.
Prima di concludere questa parte, si osservi che, nella realtà, quasi il 50% dei pazienti con 1 o 2 metastasi cerebrali non sono candidabili per l'asportazione chirurgica a causa dell'inaccessibilità delle lesioni, l'estensione della malattia sistemica ovvero per altri fattori di complicazione. A questi pazienti, e ad altri con metastasi multiple, normalmente si offre come trattamento standard la radioterapia panencefalica. Con tale terapia in effetti sino a quasi il 50% di essi ottiene un miglioramento dei sintomi neurologici e il 50-70% mostra una risposta obiettiva.
La chemioterapia raramente ha il ruolo di terapia primaria nel caso di metastasi cerebrali. Molte neoplasie che metastasizzano al cervello (per es., il carcinoma polmonare non a piccole cellule, neoplasie in cui il sito di origine primario è sconosciuto o il melanoma) sono insensibili alla terapia farmacologica o risultano già pesantemente trattate con agenti che si riteneva potenzialmente efficaci.
Per la maggior parte dei pazienti con metastasi cerebrali la mediana di sopravvivenza è di soli 4-6 mesi, dopo la radioterapia panencefalica. Tuttavia alcuni pazienti (con età inferiore a 60 anni, lesione singola e malattia sistemica sotto controllo) possono raggiungere una sopravvivenza maggiore, per il fatto che sono in grado di essere sottoposti a un approccio terapeutico più aggressivo. Ad esempio, una parte di questi pazienti riescono ad affrontare un'altra operazione chirurgica o la radiochirurgia stereotassica. Con trattamento aggressivo la mediana di sopravvivenza arriva a 40 settimane e più.
Metastasi leptomeningee

Il coinvolgimento delle leptomeningi avviene in circa il 5% dei pazienti di cancro e viene rilevato più frequentemente, mano in mano che le metodiche diagnostiche migliorano ed i pazienti vivono più a lungo. Le neoplasie di origine più comuni sono il melanoma e i carcinomi mammario e polmonare. Il cancro raggiunge le leptomeningi come risultato della propagazione di cellule cancerose attraverso il flusso sanguigno. Le cellule maligne risultano in genere disseminate per tutto il nevrasse dal flusso del liquido cerebrospinale.
I segni e i sintomi sono riferibili ad una o più delle seguenti situazioni: danno locale ai nervi che viaggiano attraverso il fluido spinale (paralisi dei nervi cranici, debolezza motoria con comparsa di dolori radicolari, parestesie, fitte); invasione diretta del cervello o dei tessuti spinali; interruzione dei vasi sanguigni diretti a quei tessuti (deficit neurologici focali o attacchi epilettici); ostruzione del normale flusso del liquido cerebrospinale (cefalea ed aumento della pressione endocranica); interferenza con il normale funzionamento del cervello (encefalopatia); ovvero infiltrazione perivascolare da parte di cellule tumorali, con conseguente ischemia locale e sintomi da colpo apoplettico.
La diagnosi si effettua con l'esame del liquido cerebrospinale e/o la risonanza magnetica del cervello e del midollo spinale. Lo studio del liquor rivela la presenza di cellule maligne nel 50% dei pazienti; tuttavia in almeno il 10% dei malati con sospetto coinvolgimento leptomeningeo l'esame citologico rimane persistentemente negativo. L'aumento del numero di punture lombari (fino a 6) e del volume di liquido rimosso (10 ml per puntura) incrementa la possibilità di diagnosi positiva. Nel liquido cefalorachidiano la concentrazione di proteine è normalmente elevata, quella di glucosio può essere bassa, con presenza di pleocitosi. Lo studio radiologico può evidenziare idrocefalo in assenza di lesione massiva o enhancement diffuso delle leptomeningi.
Senza terapia la mediana di sopravvivenza è di 4-6 settimane, con decesso dovuto a progressivo deterioramento neurologico. Spesso le metastasi leptomeningee sono una manifestazione dello stadio finale della malattia principale e la terapia sintomatica può essere la soluzione più appropriata. Corticosteroidi ed analgesici offrono un temporaneo alleviamento. Ai pazienti con malattia sistemica minimale ed accettabile condizione fisica generale può essere offerto un trattamento per attenuare i sintomi e prolungare la sopravvivenza.
La sopravvivenza mediana può essere aumentata da 3 a 6 mesi con radioterapia ai siti sintomatici e delle aree malate più voluminose individuate con lelastre, e con terapia intratecale con metotrexato, citarabina e tiotepa (effettuata con puntura lombare o catetere Ommaya).
Benché la chemioterapia riesca a prolungare significativamente la sopravvivenza di pazienti con malattia ematologica, tipo leucemia o anche linfoma, ottenere un beneficio attraverso il liquido cerebrospinale quando si ha a che fare con tumori solidi risulta perlomeno dubbio. In tali circostanze il decesso avviene per malattia sistemica avanzata.
La maggior complicanza della terapia intratecale a base di metotrexato è rappresentata da una leucoencefalopatia necrotizzante
che può svilupparsi dopo mesi di terapia in quei pochi pazienti che giovano di una sopravvivenza prolungata. Questo effetto tossico devastante è comune soprattutto nei pazienti sottoposti a radioterapia precedente o contemporaneamente alla terapia intratecale con metotrexato.
Dolore e cure terminali
Cure palliative è un tipo speciale di cura fornito per migliorare la qualità della vita dei pazienti che soffrono di una malattia grave o pericolosa per la vita, come il cancro. Lo scopo delle cure palliative non è quello di curare ma di prevenire o curare, il prima possibile, i sintomi e gli effetti collaterali della malattia e del suo trattamento, oltre ai relativi problemi psicologici, sociali e spirituali. Le cure palliative sono anche chiamate cure di comfort, cure di supporto e gestione dei sintomi.
Le cure palliative sono fornite durante l'esperienza di un paziente con il cancro. Di solito inizia alla diagnosi e continua attraverso il trattamento, le cure di follow-up e la fine della vita.
Collegamenti Web
- www.neuro-oncologia.eu – Associazione Italiana di Neuro-Oncologia
Bibliografia
- (EN) Lisa M. DeAngelis (2001). Brain Tumors., N Engl J Med. 2001 Jan 11;344(2):114-23.
- (EN) Primary and metastatic brain tumors (2010). Primary and metastatic brain tumors. In Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (2009). Cancer Management: A Multidisciplinary Approach, 12th Edition. CancerNetwork. Updated: 2010 Mar 11.
- (EN) Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007). World Health Organization Classification of Tumours of the Central Nervous System. IARC, Lyon ISBN 92-832-2430-2.
- (EN) Packer RJ (2008). Childhood brain tumors: accomplishments and ongoing challenges, su jcn.sagepub.com. URL consultato il 4 febbraio 2022 (archiviato dall'url originale l'11 aprile 2013). J Child Neurol. 2008 Oct;23(10):1122-7.
- (EN) Wen PY, Kesari S (2008). Malignant gliomas in adults. N Engl J Med. 2008 Jul 31;359(5):492-507. Erratum in: N Engl J Med. 2008 Aug 21;359(8):877.
Anatomia patologica e medicina interna
- Robbins e Cotran, Le basi patologiche delle malattie, 7ª ed., Torino-Milano, Elsevier Masson, 2008, ISBN 978-88-85675-53-7.
- Mariuzzi, Anatomia patologica e correlazioni anatomo-cliniche, Padova, Piccin, 2006, ISBN 978-88-299-1769-3.
- Harrison, Principi di Medicina Interna, 16ª ed., New York-Milano, McGraw-Hill, 2006, ISBN 88-386-2459-3.
Farmacologia
- Brunton, Lazo, Parker, Goodman & Gilman - Le basi farmacologiche della terapia, 11ª ed., McGraw Hill, 2006, ISBN 978-88-386-3911-1.
- Bertram G. Katzung, Farmacologia generale e clinica, Padova, Piccin, 2006, ISBN 88-299-1804-0.
Neurologia
- C. Loeb, E. Favale, Neurologia di Fazio Loeb, Roma, Società Editrice Universo, 2003, ISBN 88-87753-73-3.
- B. Bergamasco, R. Mutani, La neurologia di Bergamini, Torino, Cortina, 2007, ISBN 88-8239-120-5.
- Allan H. Ropper, Robert H. Brown, Adams & Victor - Principi di neurologia, Milano - New York, McGraw-Hill Companies, 2006, ISBN 88-386-3909-4.
Linee guida
- (EN) U.S. Department of Health & Human Services. National Guideline Clearinghouse (2009).[https://www.guideline.gov/search/search.aspx?term=brain+tumor¨s=1 Brain tumor]. URL consultato il 21 gennaio 2011.
- (EN) National Institute for Health and Clinical Excellence (2006).Service guidance for improving outcomes for people with brain and other central nervous system tumours. URL consultato il 21 gennaio 2011.
- (EN) American Academy of Family Physicians (2008).Primary Brain Tumors in Adults. A cura di Chandana SR, Movva S, Arora M, Singh T. URL consultato il 21 gennaio 2011.
- (EN) National Cancer Institute (2010). General Information About Adult Brain Tumors. URL consultato il 21 gennaio 2011.
Linee guida italiane
- AIOM - Associazione Italiana di Oncologia Medica (2009).Neoplasie cerebrali.A cura di Brandes AA, Calbucci F, Leonardi M, Reni M, Spagnolli F, Tosoni A, Labianca R, Ferrarese F, Carapella C. URL consultato il 21 gennaio 2011.
- Ministero della Salute. Basi Scientifiche Linee Guida (2007).Tumori cerebrali. A cura di Rosella Silvestrini. URL consultato il 21 gennaio 2011.
Voci correlate
- Classificazione dei tumori del sistema nervoso centrale
- Gradazione dei tumori del sistema nervoso centrale
- Oligodendroglioma anaplastico
- Neuroradiologia dei tumori primitivi cerebrali
- Sistema nervoso
- Tumore
Collegamenti esterni
Vengono qui riportati i link alle maggiori riviste mediche specializzate.
- Acta Neuropathologica, su springerlink.com. URL consultato il 16 gennaio 2011 (archiviato dall'url originale il 6 dicembre 2009).
- American Journal of Neuroradiology, su ajnr.org.
- Annals of Neurology, su www3.interscience.wiley.com. URL consultato il 4 febbraio 2022 (archiviato dall'url originale il 5 gennaio 2013).
- Archives of Neurology, su archneur.ama-assn.org.
- Brain, su brain.oxfordjournals.org.
- Brain Pathology, su www3.interscience.wiley.com. URL consultato il 4 febbraio 2022 (archiviato dall'url originale il 5 gennaio 2013).
- Brain Tumor Pathology, su springerlink.com. URL consultato il 4 febbraio 2022 (archiviato dall'url originale l'11 aprile 2013).
- British Journal of Neurosurgery, su informahealthcare.com.
- Child's Nervous System, su springerlink.com. URL consultato il 16 gennaio 2011 (archiviato dall'url originale il 3 ottobre 2012).
- Clinical Neuropathology, su dustri.com.
- Glia, su www3.interscience.wiley.com. URL consultato il 4 febbraio 2022 (archiviato dall'url originale il 22 aprile 2012).
- International Journal of Radiation Oncology, Biology, Physics, su sciencedirect.com.
- Journal of Neurology, su springerlink.com. URL consultato il 4 febbraio 2022 (archiviato dall'url originale l'11 aprile 2013).
- Journal of Neurology, Neurosurgery, and Psychiatry, su jnnp.bmj.com.
- Journal of Neuro-Oncology, su springerlink.metapress.com.
- Journal of Neuropathology and Experimental Neurology, su journals.lww.com.
- Journal of Neurosurgery, su thejns.org.
- The Lancet Neurology, su thelancet.com.
- Nature Reviews Neurology, su nature.com.
- Neurological Research, su ingentaconnect.com. URL consultato il 16 gennaio 2011 (archiviato dall'url originale l'11 febbraio 2011).
- Neurology, su neurology.org.
- Neuroradiology, su springerlink.com. URL consultato il 4 febbraio 2022 (archiviato dall'url originale l'11 aprile 2013).
- Neuro-Oncology, su neuro-oncology.oxfordjournals.org.
- Neurosurgery, su journals.lww.com.
- Neurosurgical Focus, su thejns.org.
- Pediatric Neurology, su journals.elsevierhealth.com.
- Pediatric Neurosurgery, su content.karger.com.
- Revista de neurologia, su revneurol.com.
- Surgical Neurology, su journals.elsevierhealth.com. URL consultato il 16 gennaio 2011 (archiviato dall'url originale il 23 luglio 2007).
Gli articoli di rilevanza in materia pubblicati altrove (Cancer, JAMA, JCO, NEJM, ecc.) sono normalmente segnalati dalla newsletter elettronica
- Current Neuro-Oncology, su brainlife.org. URL consultato il 27 giugno 2011 (archiviato dall'url originale il 25 luglio 2011).