
| Sindrome del QT lungo | |
|---|---|
 | |
| Malattia rara | |
| Specialità | cardiologia |
| Classificazione e risorse esterne (EN) | |
| ICD-10 | I49.8 |
| OMIM | 192500 |
| MeSH | D008133 |
| eMedicine | 157826 |
La sindrome del QT lungo (LQTS, dall'inglese "Long Q-T Syndrome") è una rara anomalia cardiaca caratterizzata da una ritardata ripolarizzazione delle cellule miocardiche ed associata a sincope o morte improvvisa. La LQTS può degenerare in aritmie maligne come torsioni di punta e fibrillazione ventricolare o può portare all'arresto cardiaco. Le aritmie nei pazienti affetti da LQTS sono spesso scatenate dall'esercizio fisico o dagli stimoli emotivi.
Mentre la sindrome del QT lungo è una patologia genetica nella quale il prolungamento del intervallo QT è determinato da una o più alterazioni elettrofisiologiche, l'intervallo QT misurato può risultare prolungato anche per fattori ambientali (es. disordini elettrolitici e farmaci). La diagnosi di sindrome del QT lungo deve quindi escludere le cause "acquisite" che sono modificabili e quindi potenzialmente curabili. Il prolungamento del QT acquisito può derivare dall'assunzione di alcuni farmaci, basso livello di potassio nel sangue, basso livello di calcio nel sangue o insufficienza cardiaca. I farmaci implicati includono alcuni antiaritmici, antibiotici, antipsicotici, metadone e diverse classi di antidepressivi. (SSRI, SNRI, TCA, mirtazapina e trazodone)
La diagnosi si basa sulla misura dell'intervallo QT all'elettrocardiogramma (ECG) insieme ai riscontri obiettivi. L'intervallo QT si misura dall'inizio dell'onda Q al termine dell'onda T. Tale misura (espressa normalmente in millisecondi) deve poi essere corretta per la frequenza cardiaca al momento della misurazione. Si definisce così il "QT corretto" o QTc. Vi sono numerose formule di correzione che possono essere utilizzate ma nalla pratica clinica si usa prevalentemente la correzione di Bazet.
La gestione della patologia può includere la prevenzione di un intenso esercizio fisico, l'uso di beta-bloccanti o un defibrillatore cardiaco impiantabile. Per le persone con LQTS che sopravvivono all'arresto cardiaco e non sono trattate per LQTS, il rischio di morte entro 15 anni è superiore al 50%. Con un trattamento adeguato questo diminuisce a meno dell'1% in 20 anni.
Si stima che la sindrome del QT lungo colpisca 1 su 2000 persone. La patologia è trasmessa in oltre il 90% dei casi con una modalità autosomica dominante ma l'espressitivtà della malattia (cioè la severità delle manifestazioni cliniche) è tendenzialmente più elevata nelle femmine le quali hanno un intervallo QT fisiologicamente più lungo rispetto ai maschi. La maggior parte delle persone con questa condizione sviluppa sintomi prima dei 40 anni. È una causa relativamente comune di morte improvvisa insieme alla sindrome di Brugada e alla displasia ventricolare destra aritmogena. Negli Stati Uniti provoca circa 3.500 morti all'anno. La condizione fu descritta per la prima volta nel 1957.
Storia
Fu riscontrata per la prima volta nel 1957 da Jervell e Lange-Nielsen, che descrissero la variante della sindrome associata a sordità neurologica congenita. I due autori notarono che gli episodi di morte improvvisa avevano carattere familiare: si trasmetteva ai discendenti. Successivamente, nel 1958, Levine e Woodworth osservarono una morte improvvisa di un tredicenne con i sintomi tipici della malattia. Negli anni '60 due studi indipendenti, l'uno condotto da Cesarino Romano (un pediatra italiano) nel 1963 e l'altro da Owen Conor Ward (un pediatra irlandese) nel 1964, contribuirono alla definizione di una sindrome congenita ereditata secondo il modello autosomico dominante, successivamente denominata Sindrome di Romano-Ward, caratterizzata da sincopi ed anomalie dell'ECG, senza deficit uditivo. L'istituzione del registro internazionale per la sindrome del QT lungo (LQTS Registry) nel 1979 ha permesso di valutare numerose famiglie colpite dalla patologia a livello mondiale. Ciò ha aiutato a rilevare molti dei numerosi geni coinvolti.
Segni e sintomi
Molte persone con sindrome del QT lungo non hanno segni o sintomi.
I sintomi che si verificano sono generalmente causati da ritmi cardiaci anormali o aritmie, più comunemente una forma di tachicardia ventricolare polimorfa chiamata torsades de pointes. Se l'aritmia è transitoria e ritorna da sola a un ritmo sinusale, il paziente può avere manifestato una sincope. Tuttavia, se l'aritmia persiste, a causa di una mancata circolazione sanguigna adeguata la persona può arrivare all'arresto cardiaco, che porta alla morte improvvisa.
Le aritmie che portano a svenimenti e morte improvvisa hanno maggiori probabilità di verificarsi in risposta a circostanze specifiche, in parte determinato da quale variante genetica è responsabile della condizione. Mentre le aritmie possono verificarsi in qualsiasi momento, in alcune forme di LQTS le aritmie sono più comunemente osservate in risposta all'esercizio fisico o allo stress mentale (LQT1), in altre forme a seguito di un improvviso rumore forte (LQT2) e in alcune forme durante il sonno o immediatamente dopo il risveglio (LQT3).
Alcune forme rare di sindrome del QT lungo sono associate a sintomi che colpiscono altre parti del corpo. Questi includono la sordità nella variante di Jervell e Lange-Nielsen e la paralisi periodica nella forma di Andersen-Tawil (LQT7).
Rischio di aritmie
Se i soggetti con sindrome del QT lungo hanno un aumentato rischio di sviluppare ritmi cardiaci anormali rispetto a quelli senza la condizione, in realtà il rischio assoluto di aritmie è molto variabile. Il predittore più forte che ci permette di dire se un soggetto svilupperà torsioni di punta (TdP) è se in passato ha già avuto episodi di TdP spontaneo o un'altra forma di arresto cardiaco. Anche se non è stata osservata un'aritmia, una persona con LQTS che ha in anamnesi una sincope è a rischio di aritmie, poiché verosimilmente la sincope in questi casi è spesso dovuta a un'aritmia auto-terminante non documentata.
Le due variabili cliniche con maggiore correlazione con il rischio aritmico sono la durata del QTc ed il genotipo. COmbinando tali variabili è possibile stimare in modo preciso il rischio di eventi aritmici e modulare l'approccio terapeutico/preventivo.
Epidemiologia
Fra i soggetti affetti, l'incidenza di torsione di punta, sincope e morte cardiaca improvvisa è più elevata in caso di sordità congenita, precedente tachiaritmia o sincope; la stessa aumenta inoltre proporzionalmente all'allungamento dell'intervallo QT. Si calcola che il rischio relativo di aritmie maligne aumenti di 1,1 - 1,2 volte per ciascun prolungamento di 10 msec del QTc oltre i valori normali. Si stima che l'ereditarietà di LQTS colpisca tra uno su 2.500 e uno su 7.000 persone.
Nei soggetti geneticamente predisposti, costituiscono fattori di rischio per l'insorgenza di aritmie maligne gli incrementi improvvisi del tono simpatico, come può accadere in concomitanza di sforzi eccessivi o emozioni violente.
Varianti cliniche
Si distinguono due diverse sindromi congenite caratterizzate da allungamento dell'intervallo QT e rischio di morte improvvisa per aritmie ventricolari:
- Sindrome di Romano-Ward, che viene ereditata secondo il modello autosomico dominante (non associata a sordità neurologica congenita o ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza).
- Sindrome di Jervell-Lange-Nielsen, che viene ereditata secondo il modello autosomico recessivo (associata a sordità neurologica congenita o ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza).
Eziologia
Nella LQTS si distinguono due forme: congenita ed acquisita.
Forme acquisite
VI sono numerosi farmaci che possono prolungare l'intervallo QT e provocare la forma aquisita di QT lungo. Tali farmaci sono chiaramente contrindicati nei pazienti affetti da LQTS.
La maggior parte dei casi riscontrati nella pratica clinica riguarda forme acquisite, che possono essere suddivise in due categorie: quelle generate da disturbi dell'equilibrio idro-elettrolitico e quelle dipendenti dalla somministrazione di farmaci.
- disordini elettrolitici:
- forme indotte da farmaci:
- farmaci antiaritmici
- Anti-istaminici
-
Antibiotici
- Macrolidi (Eritromicina)
- Alcuni fluorochinolonici
- Ansiolitici maggiori
- Antidepressivi triciclici
- Agenti attivi sulla motilità gastrointestinale
- Antipsicotici
- Metadone
Proprio come le forme congenite di LQTS, le forme acquisite possono condurre ad aritmie mortali. Il trattamento consiste nella correzione dello squilibrio elettrolitico, nella risoluzione della sua causa e nella sospensione della terapia con il farmaco indiziato dell'allungamento del QT.
Dato il largo impiego, la tendenza ad interazioni con altri farmaci e la insita capacità di determinare allungamento dell'intervallo QT, l'eritromicina è probabilmente la causa prevalente di sindrome acquisita del QT lungo. Infatti, l'uso di eritromicina è associato con un'incidenza di morte improvvisa più che doppia rispetto ad altri antibiotici
In aggiunta alle due maggiori categorie elencate sopra, deve essere ricordato che esistono altre cause di prolungamento dell'intervallo QT come l'anoressia nervosa, l'ipotiroidismo, l'infezione da HIV, la miocardite e l'infarto miocardico.
Forme congenite
Le forme congenite di LQTS possono essere determinate da mutazioni di uno di diversi geni finora identificati. Queste mutazioni tendono a prolungare la durata del potenziale d'azione ventricolare (APD), allungando in tal modo l'intervallo QT. Le forme congenite possono essere ereditate come carattere autosomico dominante o autosomico recessivo. La forma autosomica recessiva è associata ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza (LQTS8) o a sordità neurologica congenita (LQTS1). Un numero sempre crescente di loci genici viene identificato in associazione alle LQTS. I test genetici per le LQTS è disponibile nella pratica clinica e può essere di aiuto anche per impostare la terapia appropriata (Overview of LQTS Genetic Testing). Le più comuni cause di LQTS riguardano mutazioni nei geni KCNQ1 (LQTS1), KCNH2 (LQTS2) e SCN5A (LQT3); il seguente è un elenco di tutti i geni finora identificati nella genesi della LQTS:
| Tipo | Mutazione | Note |
| LQTS1 | subunità alfa del canale del potassio a rettificazione lenta ritardata (KvLQT1 o KCNQ1) | La corrente attraverso il canale eteromerico (KvLQT1 + minK) è nota come IKs. Queste mutazioni causano LQTS mediante una riduzione dell'intensità di corrente attraverso il canale ionico. Questa corrente di ripolarizzazione è determinante nel riportare il potenziale cellulare verso il valore di riposo; con la sua diminuzione si determina un incremento della durata del potenziale d'azione (APD). Queste mutazioni sono le più frequenti ma corrispondono ad un quadro clinico meno severo. Mutazioni in omozigosi di questo gene provocano la forma di LQTSassociata a sordità congenita (sindrome dei Jervell e Lange-Nielsen) |
| LQTS2 | subunità alfa del canale a rettificazione ritardata rapida (HERG + MiRP1) | La corrente attraverso questo canale è nota come IKr. Anche questo fenotipo è probabilmente causato da una riduzione della corrente ripolarizzante. |
| LQTS3 | subunità alfa del canale del sodio (SCN5A) | La corrente attraverso questo canale è comunemente indicata come INa. Si ritiene che la corrente depolarizzante attraverso il canale nella fase tardiva del potenziale d'azione conduca al suo prolungamento. La corrente tardiva è dovuta alla tardiva inattivazione del canale. Di conseguenza esso può entrare in uno stato di attivazione, durante il quale un flusso significativo di corrente continua ad entrare nella cellula. Le mutazioni di questo canale, sebbene meno comuni, si traducono in un fenotipo caratterizzato da una maggiore mortalità. |
| LQTS4 | proteina d'ancoraggio Anchirina B | LQTS4 è molto rara. L'anchirina B assicura i canali ionici alla membrana cellulare. |
| LQTS5 | beta subunità MinK (o KCNE1) che si prende parte all'assemblaggio del canale ionico per IKs assieme a KvLQT1 | - |
| LQTS6 | beta subunità MiRP1 (o KCNE2) che assieme a HERG prende parte all'assemblaggio del canale ionico per IKr | - |
| LQTS7 | canale del potassio KCNJ2 (o Kir2.1) | La corrente attraverso questo canale e KCNJ12 (Kir2.2) è chiamata IK1. LQTS7 ha come espressione la sindrome di Andersen-Tawil. |
| LQTS8 | subunità del canale del calcio Cav1.2 codificata dal geneCACNA1c. | La LQTS8 corrisponde alla sindrome di Timothy ed è associata ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza. |
| LQTS9 | Caveolin 3 | |
| LQTS10 | SCN4B | |
| LQTS11 | AKAP9 | |
| LQTS12 | SNTA1 |
- LQTS1
La LQTS1 è la forma più comune di LQST, rappresentando il 30-35% di tutti i casi. Il gene responsabile della LQTS1 è il KCNQ1, che è stato identificato sul cromosoma 11 nel locus p15.5. KCNQ1 codifica il canale del potassio voltaggio-dipendente KvLQT1, ampiamente espresso nel cuore. Si ritiene che il prodotto del gene KCNQ1 produca la subunità alfa che interagisce con altre proteine (particolarmente con la subunità beta minK) per costituire il canale ionico della corrente IKs, che è responsabile della corrente di rettificazione ritardata lenta del potassio nel potenziale d'azione cardiaco.
Le mutazioni al gene KCNQ1 possono dare luogo ad espressioni fenotipiche secondo il modello autosomico dominante oppure autosomico recessivo all'interno della stessa famiglia. In caso di sindrome clinica espressa, ereditata in modo autosomico recessivo, mutazioni omozigoti nel gene KVLQT1 conducono ad un marcato prolungamento dell'intervallo QT (a causa della pressoché completa perdita della corrente IKs) e sono associate a sordità neurologica congenita.
La gran parte degli individui affetti da LQTS1 mostra un prolungamento paradossale dell'intervallo QT con l'infusione di adrenalina.
Sono state identificate molte mutazioni missense del gene LQTS1. Queste sono meno spesso associate a morte improvvisa rispetto alla LQTS2.
- LQTS2
Il tipo LQTS2 è la seconda variante genica più comune, rappresentando il 25-30% di tutti i casi. Questa forma di LQTS molto probabilmente coinvolge mutazioni del gene human ether-a-go-go related gene (HERG), il cui locus è localizzato sul cromosma 7. Il gene HERG (noto anche come KCNH2) è parte della componente rapida della corrente di rettificazione rapida del potassio (IKr). La corrente IKr è principalmente responsabile del ritorno del potenziale d'azione cardiaco al livello precedente la depolarizzazione, e pertanto della lunghezza dell'intervallo QT. Il gene HERG normalmente funzionante protegge dalle depolarizzazioni post-potenziali precoci (EADs).
- LQTS3
La LQTS3 è determinata da mutazioni del gene, che codifica la subunità alfa del canale del Na+. Questo gene è localizzato sul cromosoma 3p21-24, ed è noto come SCN5A (anche hH1 e NaV1.5). Le mutazioni implicate nella LQTS3 determinano un prolungamento dell'influsso di Na+ durante la depolarizzazione perché, paradossalmente, il canale del sodio mutante si inattiva più rapidamente e può aprirsi ripetutamente durante il potenziale d'azione.
Un gran numero di mutazioni sono state caratterizzate come determinanti o predisponenti alla LQTS3. È stato suggerito che il calcio possa essere un agente regolatore del canale SCN5A, e che gli effetti del calcio sul canale SCN5A possano spiegare il meccanismo mediante il quale alcune mutazioni causano la LQTS3. Inoltre, mutazioni a carico di SCN5A possono causare la sindrome di Brugada, malattia del sistema cardiaco di conduzione e cardiomiopatia dilatativa.
- LQTS5
È una forma autosomica dominante relativamente poco comune di LQTS. Essa sottintende mutazioni nel gene KCNE1, che codifica la subunità beta del canale del potassio MinK. Nelle sue rare forme omozigoti può potare alla sindrome di Jervell e Lange-Nielsen.
- LQTS6
È una forma autosomica dominante relativamente poco comune di LQTS. Essa sottintende mutazioni nel gene KCNE1, che codifica la subunità beta del canale del potassio MiRP1, che costituisce una parte della corrente di ripolarizzazione IKr.
- LQTS7
La sindrome di Andersen-Tawil è una forma autosomica dominante di LQTS associata con deformazioni scheletriche. Essa è causata da mutazioni del gene KCNJ2, che codifica la proteina Kir 2.1 i un canale del potassio. La sindrome è caratterizzata da LQTS, paralisi periodica e anomalia dello sviluppo scheletrico, come clinodattilia, attaccatura bassa delle orecchie e micrognazia. Le manifestazioni sono estremamente variabili.
- LQTS8
La sindrome di Timothy è imputabile a mutazioni nel canale del calcio Cav1.2 codificato dal gene CACNA1c. Dal momento che il canale del calcio Cav1.2 abbonda in molti tessuti, i pazienti affetti da sindrome di Timothy presentano anche altre manifestazioni cliniche, fra le quali altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza.
- LQTS9
La variante recentemente scoperta è causata da mutazioni in una proteina strutturale della membrana plasmatica, la caveolina-3. Le caveoline formano uno specifico dominio della membrana chiamata caveola, nella quale fra le altre ha sede il canale voltaggio-dipendente per il sodio NaV1.5. Come avviene nella LQTS3, queste particolari mutazioni incrementano la cosiddetta corrente tardiva del sodio, che perturba la ripolarizzazione cellulare.
- LQTS10
Questa nuove mutazione genica in causa nella LQTS risiede nel gene SCN4B, che codifica la proteina NaVβ4, una subunità ausiliaria alla subunità NaV1.5 (gene: SCN5A) che costituisce il poro del canale del sodio voltaggio-dipendente nel cuore. La mutazione conduce ad un incremento dei valori di potenziale di membrana, in corrispondenza del quale scatta l'inattivazione della corrente del sodio. Fino a questo momento è stata identificata una sola mutazione in un solo paziente.
Sindrome di Jervell e Lange-Nielsen
È la forma autosomica recessiva di LQTS. È associata a sordità neurologica congenita o ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza. Essa è causata specificatamente da una mutazione dei geni KCNE1 e KCNQ1. Degli individui non trattati, circa il 50% muore entro l'età di 15 anni a causa di aritmie ventricolari.
Sindrome di Romano-Ward
La sindrome di Romano-Ward è la forma autosomica dominante di LQTS. Non è associata a sordità neurologica congenita o ad altre malattie cardiache congenite, autismo, sindattilia completa e immunodeficienza.
Fisiopatologia
Alla base di tutte le forme di LQTS c'è una anormale ripolarizzazione del miocardio. Le anomalie della ripolarizzazione causano differenze nella refrattarietà dei miocardiociti. A causa di queste differenze, eventuali post-depolarizzazioni (che si verificano più di frequente nei pazienti affetti da LQTS) possono propagarsi alle cellule contigue, conducendo ad aritmie ventricolari da rientro.
Si crede che le cosiddette depolarizzazioni post-potenziali precoci (EADs) che si osservano nelle LQTS siano dovute ad una riapertura dei canali del calcio di tipo L quando ancora si è durante il plateau del potenziale di azione cardiaco: ciò in virtù dell'aumento della durata del potenziale (che è alla base di questa sindrome) e della refrattarietà TEMPO dipendente che caratterizza specificatamente i canali L. Dal momento che la stimolazione adrenergica può esaltare l'azione di questi canali, condizioni come l'esercizio fisico e lo stress emotivo possono favorire le aritmie. Di norma, durante attivazione adrenergica, le correnti di ripolarizzazione sono amplificate ed accorciano il potenziale d'azione. In assenza di accorciamento del potenziale d'azione, ed in presenza di un incremento della corrente attraverso i canali L del calcio, le EADs possono manifestarsi.
Si ritiene che le cosiddette depolarizzazioni post-potenziali tardive (DADs) siano dovute ad un incremento della concentrazione di Ca2+ nel reticolo sarcoplasmatico. Questo sovraccarico può causare ondate di rilascio spontaneo nel citoplasma di Ca2+ durante la ripolarizzazione; il Ca2+ abbandonerebbe poi la cellula attraverso lo scambiatore 3Na+/Ca2+, con una netta corrente di depolarizzazione.
Diagnosi
La diagnosi di LQTS non è facile dal momento che il 2.5% della popolazione sana ha un QT prolungato, e molti portatori di mutazioni associate alla LQTS presenta QT normale. Tale fenomeno, comune a molte malattie genetiche, è definito come penetranza incompleta, per la LQTS può dipende dal genotipo e può arrivare anche al 50% nella variante LQT5. La penetranza media della LQTS è del 60% circa (corrispondente al 40% di portatori di mutazioni con QT normale). Le varianti LQT1, LQT3 ed LQT8 presentano penetranza elevata (> 70%).
Secondo le attuali linee guida della Società Europea di Cardiologia la diagnosi di LQTS si pone con i seguenti Criteri:
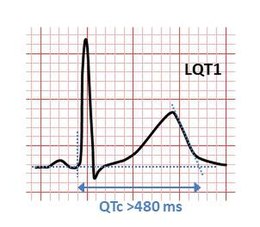
Presenza di QTc ≥ 480 ms in ripetuti ECG con o senza storia di episodi sincopali
Presenza di Score diagnostico > 3
Presenza di QTc 460 ms ÷ 480 ms in pazienti con sincope ed in assenza di cause secondarie di prolungamento del QT
Presenza di una mutazione patogenica nota indipendentemente dalla durata del QT
Lo "score" diagnostico è stato proposto in affiancamento a criteri principali per i casi dubbi. Il punteggio è calcolato assegnando punti in base a vari criteri elencati qui di seguito. Con 4 o più punti la probabilità di LQTS è alta, e con 1 punto o meno la probabilità è bassa; 2 o 3 punti indicano una probabilità intermedia.
- QTc (definito come intervallo QT/radice quadrata dell'intervallo RR)
- >= 480 msec - 3 punti
- 460-470 msec - 2 punti
- 450 msec e genere maschile - 1 punto
- Tachicardie ventricolari tipo torsades de pointes - 2 punti
- Alternanza dell'onda T - 1 punto
- Avvallamento dell'onda T in almeno 3 derivazioni all'ECG- 1 punto
- Bassa frequenza cardiaca per l'età (bambini) - 0.5 punti
- Sincope (non possono essere assegnati punti sia alla sincope che alle torsioni di punta allo stesso soggetto)
- In caso di stress - 2 punti
- Al di fuori di condizioni di stress - 1 punto
- Sordità congenita - 0.5 punti
- Storia familiare (lo stesso membro della famiglia non può essere conteggiato sia per la morte improvvisa sia per la LQTS)
- Altri membri della famiglia con diagnosi sicura di LQTS - 1 punto
- Morte improvvisa nei familiari stretti (membri con età inferiore a 30 anni) - 0.5 punti
Terapia
Nei pazienti asintomatici, senza dimostrazione di aritmie ventricolari ed in assenza di anamnesi familiare positiva per morte improvvisa, è raccomandata la sola osservazione ed eventualmente la terapia farmacologica senza necessità di arrivare al dosaggio massimo tollerato.
Nelle LQTS1 e LQTS2 possono essere utilizzati i betabloccanti; mentre nelle LQTS 3 sono preferibili gli anti-aritmici di classe Ib, come la mexiletina. Gli stessi farmaci possono essere adoperati nel trattamento dei pazienti in urgenza, con l'accortezza di utilizzare soltanto la lidocaina fin quando non venga confermata la diagnosi di LQTS1 o LQTS2, poiché i betabloccanti possono peggiorare le aritmie nei pazienti portatori di LQTS3.
La terapia anti-aritmica deve essere, invece, adottata e condotta al dosaggio massimo tollerato in quei paziente asintomatici ma con evidenza di aritmie ventricolari non sostenute e storia familiare di morte improvvisa. In questi pazienti non è strettamente raccomandato l'impianto del defibrillatore cardiaco impiantabile (ICD). Quest'ultimo è, invece, assolutamente indicato (indicazione di classe I) nei pazienti con sincope o che abbiano già avuto un episodio di arresto cardiaco.
La funzione di pace-maker del dispositivo deve essere sfruttata in coloro che presentano aritmie in occasione di bradicardia o pause del ritmo cardiaco. Nei pazienti che presentino ancora dei sintomi, nonostante la terapia medica ottimale, è indicato l'intervento chirurgico di gangliectomia cervico-toracica sinistra, con distruzione del ganglio stellato e dei primi tre o quattro gangli simpatici toracici.
Prognosi e stratificazione del rischio
Per i pazienti affetti da LQTS e non trattati il rischio di incorrere in un evento (sincope o arresto cardiaco) può essere stimato conoscendo il loro genotipo (LQTS1-10), dal genere e dall'intervallo QTc.
- Rischio elevato (>50%)
QTc>500 msec LQTS1 & LQTS2 & LQTS3 (maschi)
- Rischio intermedio (30-50%)
QTc>500 msec LQTS3 (femmine)
QTc<500 msec LQTS2 (femmine)& LQTS3
- Rischio basso (<30%)
QTc<500 msec LQT1 & LQT2 (maschi)
Bibliografia
- Michael H Crawford, Diagnosi e terapia in cardiologia, Milano, McGraw-Hill, 2006, ISBN 88-386-3915-9.
- Eugene Braunwald, Malattie del cuore (7ª edizione), Milano, Elsevier Masson, 2007, ISBN 978-88-214-2987-3.
- Aldo Zangara, Terapia medica ragionata delle malattie del cuore e dei vasi, Padova, Piccin, 2000, ISBN 88-299-1501-7.
- Hurst, Fuster, Alexander, O'Rourke, Il Cuore (11ª edizione), Milano, McGraw-Hill, 2005, ISBN 88-386-2998-6.
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su sindrome del QT lungo
Wikimedia Commons contiene immagini o altri file su sindrome del QT lungo