
| Sindrome di McCune-Albright-Sternberg | |
|---|---|
 | |
| Malattia rara | |
| Cod. esenz. SSN | RNG060 |
| Specialità | genetica clinica |
| Classificazione e risorse esterne (EN) | |
| ICD-9-CM | 756.54 |
| ICD-10 | Q78.1 |
| OMIM | 174800 |
| MedlinePlus | 001217 |
| eMedicine | 127233 |
| Sinonimi | |
| Displasia fibrosa monostotica Displasia fibrosa poliostotica Osteite fibrosa disseminata Sindrome di McCune-Albright Morbo di Weil-Albright | |
| Eponimi | |
| Carl Sternberg Donovan James McCune Fuller Albright A. Weil | |
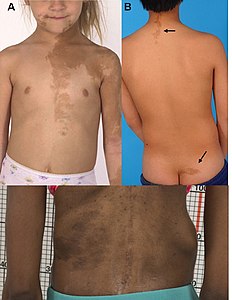
La sindrome di McCune-Albright-Sternberg (o sindrome fibrosa poliostosica) è una patologia complessa caratterizzata dalla contemporanea presenza di pseudopubertà precoce, displasia scheletrica e macchie caffellatte.
La sua esatta incidenza è ignota. Si manifesta per lo più nelle femmine (3:2 coi maschi) o in soggetti con neoplasie endocrine isolate.
Segni e sintomi
La sindrome è caratterizzata da
- displasia fibrosa poliostotica, cioè sostituzione di tessuto osseo con tessuto fibroso
- macchie caffellatte
- pseudopubertà precoce GnRH indipendente
Può essere associata ad anomalie cardiovascolari, epatobiliari, gigantismo, iperfosfaturia con ipofosforemia, ipertiroidismo, mixomi intramuscolari, osteomalacia o rachitismo, sindrome di Cushing.
Eziologia
La patologia è secondaria alla presenza di una mutazione che attiva il gene GNAS1, dovuta a mutazione somatica, più spesso, post-zigotica, con distribuzione a mosaico.
Tale mutazione riguarda il codone 201 nell'esone 8, che codifica per la subunità alfa della proteina Gs, proteina che agisce come mediatore di molti ormoni (LH, FSH, GHRH, ACTH, CRF, TSH, vasopressina, catecolamine, glucagone, PTH, calcitonina).
La varietà e la complessità della sindrome varia a seconda della precocità dell'insorgenza della mutazione durante l'embriogenesi.
Diagnosi
Viene confermata dalla valutazione della concentrazione plasmatica di estradiolo, da ecografie pelviche, RX dello scheletro, TC del cranio, studio molecolare del gene.
Terapia
Nelle femmine medrossiprogesterone, così da sopprimere la steroidogenesi gonadica, e testolattone, che inibisce la maturazione scheletrica. Inoltre ovariectomia in caso di progressione rapida e LHRH agonisti, se si associa una pubertà precoce vera.
Nei maschi ketoconazolo, inibitore della steroidogenesi gonadica e surrenale.
Per contrastare la displasia ossea si somministrano bifosfonati, inibitori degli osteoclasti. Per le deformità osse importanti si ricorre alla chirurgia.
Voci correlate
Collegamenti esterni
- (EN) Sindrome di McCune-Albright-Sternberg, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.