
| Sindrome di Prader-Willi | |
|---|---|
 | |
| Malattia rara | |
| Cod. esenz. SSN | RN1310 |
| Specialità | genetica clinica, pediatria e neurologia |
| Classificazione e risorse esterne (EN) | |
| OMIM | 176270 |
| MeSH | D011218 |
| MedlinePlus | 001605 |
| eMedicine | 947954 |
| GeneReviews | Panoramica |
| Eponimi | |
| Andrea Prader Heinrich Willi | |
La sindrome di Prader Willi (abbreviato PWS: Prader Willi Syndrome) è una malattia genetica rara (colpisce 1 su 15.000-25.000 nati vivi), caratterizzata dall'alterazione del cromosoma 15. Prende il nome dai primi che la individuarono nel 1956: Andrea Prader, Heinrich Willi, Alexis Labhart, Andrew Ziegler, e Guido Fanconi presso la Clinica pediatrica universitaria di Zurigo in Svizzera. I disturbi si presentano dalla prima infanzia, e vanno via via alternandosi, dovuti principalmente a disturbi ormonali che causano una preoccupante obesità in adolescenza.
Eziologia
La Prader-Willi è la più comune tra le sindromi di micro-delezione cromosomica. Avviene per due diverse cause accertate, entrambe di tipo genetico:
- Delezione di una regione, totale o parziale, sul cromosoma 15 di origine paterna. Questa particolare regione è sottoposta ad imprinting parentale e risulta attiva nel cromosoma paterno mentre è inattiva in quello materno.
- Disomia uniparentale materna: presenza di due copie di origine materna su entrambi i cromosomi 15, risultando entrambe inattive anche se possono essere uguali o diverse.
Delezione da imprinting
Cos'è l'imprinting
L'imprinting è un fenomeno fisiologico delle cellule somatiche. In una cellula somatica si hanno 46 cromosomi 'uguali' a due a due, quindi due copie di ogni gene, di cui ognuna proviene da un solo genitore. Per ogni gene abbiamo una copia proveniente dal padre e una dalla madre. Queste sono leggermente diverse tra di loro, come fenomeno del polimorfismo, ma codificano entrambe per la stessa proteina. Dato che della maggior parte dei geni è sufficiente una sola copia, avviene questo processo: l'imprinting silenzia uno dei due geni (viene compattato con metilazione in modo da non dare accesso alle proteine trascrizionali), e quello che viene attivamente trascritto è solo uno, che proviene a caso o dalla madre o dal padre. Questo fenomeno avviene nel corso della gametogenesi. La maggior parte dei geni, immediatamente dopo la fecondazione (stadio post-zigotico), subisce un'ondata di demetilazione che interessa la quasi totalità del genoma. I geni sottoposti ad imprinting vengono esclusi da questo fenomeno, proprio perché la metilazione in questo caso serve ad indicare la provenienza parentale del gene.
Il caso patologico della PWS
Nella PWS il gene materno è silenziato perché sotto imprinting, mentre quello paterno è deleto. Il gene in questione è del cromosoma 15, nella regione 15q11-q13. In questa patologia viene quindi a mancare il contributo paterno, e si avranno una serie di disturbi derivati dalla mancanza della/e proteina/e da esso derivanti. La PWS è strettamente correlata con la Sindrome di Angelman (AS), che è causata da imprinting paterno e delezione del gene materno. I sintomi sono molto diversi, e ci sono degli studi al riguardo, per definire come la provenienza venga identificata e possa variare radicalmente.

Il locus 15q11-q13
Questo locus contiene geni che vengono messi sotto imprinting specifico materno o paterno e del quale uno solo viene espresso. La regione deleta contiene le informazioni codificanti per la proteina umana necdin o necdina (NDN locus), vicina alla regione centromerica della delezione, tra i due geni ZNF127 e SNRPN, entrambi sotto imprinting. La necdina è una proteina nucleare che viene espressa solo da alcuni neuroni del cervello (SNC). Pare (dagli studi tuttora in corso sui topi) che governi l'arresto permanente della crescita cellulare dopo il periodo embrionale mitotico, durante il periodo di assestamento neuronale. Nonostante le precedenti ricerche di mappaggio genetico della necdina la individuassero nel cromosoma 7, si è dimostrato che l'espressione è limitata alla presenza dell'allele paterno nell'RNA del cervello neonatale. L'espressione non è riscontrata solo nel cervello, ma anche in altri tessuti, con però i maggiori livelli nella placenta e nel sistema nervoso centrale. NDN è espresso esclusivamente dall'allele paterno nei fibroblasti umani. Queste osservazioni portano a chiarire come la perdita dell'allele paterno possa dar luogo ai disordini neurologici degli individui con PWS.
Aggiornamenti sulle ricerche
Recenti ricerche puntano a capire come la metilazione distingua gli alleli materni e paterni. I geni SNRPN, MKRN3 e NDN son stati individuati e studiati ed è risultato che siano espressi solo dall'allele ereditato dal padre. Quindi i pazienti con PWS mancano dei suddetti.
- SNRP è coinvolto nello splicing del pre-mRNA (trascritto primario);
- MKRN3 codifica per una proteina zinc finger;
- NDN (vedere sopra)
Tutti questi geni hanno un'isola CpG al 5', che non è metilata nell'allele paterno espresso, mentre viene metilata in quello sotto imprinting (materno). Una scoperta importante è che il segnale di imprinting per il gene SNRP inizia già nella gametogenesi maschile e femminile. Ulteriori informazioni al riguardo:[1]
Disomia uniparentale
La seconda causa è detta disomia uniparentale cioè quando la coppia dei cromosomi 15 non è più composta da un membro con materiale materno e da uno con materiale paterno bensì da 2 membri materni o paterni, perdendo di conseguenza tutto il patrimonio genetico di tipo paterno o materno. Nel caso di questa sindrome, è il cromosoma 15 materno ad essere presente in duplice copia, mentre se si avesse una duplice copia del cromosoma 15 paterno, allora si avrebbe la sindrome di Angelman.
Analisi di laboratorio
L'analisi di laboratorio viene effettuata in caso di:
- Coppie che hanno un figlio con PWS o AS e ne aspettano un altro
- Sospetti durante la gestazione (vedere in Sintomi)
- Sintomi evidenti del neonato o del bambino
Da un campione di sangue, vengono messi in coltura i linfociti, per poi essere analizzati tramite lisi cellulare in soluzione ipotonica e isolamento del DNA.
I test di biologia molecolare si sono rivelati molto utili ai fini di una corretta diagnosi precoce e allo scopo di instaurare la terapia più adatta.
Test di metilazione
Viene effettuato un Southern Blot che consiste nell'ibridare il DNA del paziente (dopo trattamento con enzimi di restrizione) con DNA "sonda" marcato nelle zone critiche da evidenziare. Se l'ibridazione ovvero l'appaiamento tra il DNA sonda e quello del paziente è pressoché perfetta, il paziente non è affetto; se invece presenta malappaiamenti, significa che nel paziente ci sono mutazioni geniche. Questa tecnica non distingue le varie mutazioni, ma è la tecnica preferita perché più economica di altre e permette di diagnosticare/escludere la PWS al 100%.
MSPCR
Metylation Specific PCR è una tecnica più specifica per il riconoscimento della PWS. L'imprinting è un compattamento di zone specifiche dovuto a metilazione delle citosine. Con questo test il DNA viene trattato con bisolfito di sodio, che muta solo le citosine non metilate in uracile. Un ciclo di PCR (duplicazione selettiva di frammenti di DNA), con primer che distinguono in metilati e non.
FISH
Ibridazione fluorescente in situ (Fluorescent In Situ Hybridization o FISH): permette di esaminare esattamente il sito compromesso mediante l'utilizzo di sonde specifiche per la regione coniugate con molecole fluorescenti. L'ibridazione di queste sonde (individuo normale) o la non ibridazione (individuo con patologia) viene rilevata mediante microscopio ottico a fluorescenza.
Sintomatologia
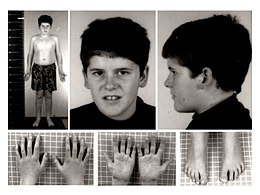
I nati con questa sindrome presentano subito una ipotonia marcata che va via via scomparendo con l'età adolescenziale. Successivamente, dai due ai sei anni, questi bambini possono sviluppare un appetito insaziabile dovuto ad una disfunzione dell'ipotalamo che li accompagnerà per tutta la loro vita. Alcuni di loro presentano un ritardo mentale che può essere lieve o grave a seconda dell'individuo, ipogonadismo, strabismo, mani e piedi piccoli. L'iperfagia però è il problema più grave; infatti, se non controllata con un regime di dieta ferrea, può portare ad obesità grave, con tutti i problemi che da essa ne derivano (vascolari, diabete ecc.) fino a compromettere la salute del soggetto stesso. I bambini PW vengono aiutati con la somministrazione dell'ormone GH della crescita che fa in modo di dare più vitalità, di farli crescere in maniera corretta e di limitare le disfunzioni metaboliche che li spingono ad aumentare di peso più facilmente rispetto ai soggetti sani.
Sintomi generali
Sintomi descritti da Prader e aa. nel 1956:
- diminuita attività fetale
- grave ipotonia infantile
- problemi alimentari nell'infanzia
- ipogonadismo ipogenitalismo
- ritardata età ossea e bassa statura
- mani e piedi piccoli
- ritardo mentale di medio grado
- facies caratteristica
- obesità (prima infanzia)
- problemi comportamentali (adolescenza)
- tendenza a sviluppare diabete (adolescenza)
Sintomi maggiori
- Ipotonia centrale neonatale e infantile con suzione debole che migliora con l'età.
- Problemi alimentari nella prima infanzia, scarso accrescimento ponderale.
- Obesità centrale dopo l'anno ma prima dei 6 anni di età.
- Tratti somatici caratteristici: dolicocefalia nell'infanzia, diametro bifrontale ristretto, occhi a mandorla, bocca piccola con labbro superiore sottile, angoli della bocca rivolti verso il basso (almeno 3 segni), rima palpebrale rivolta verso il basso.
- Ipogonadismo
- Ritardato sviluppo psicomotorio prima dei 6 anni di età;
- ritardo mentale da lieve a medio o disturbi dell'apprendimento in bambini di età maggiore. (ritardo mentale: 63% lieve, 31% medio, 6% grave)
- Iperfagia/furto del cibo/ossessione per il cibo.
- Anomalie citogenetiche o molecolari all'analisi della regione 15q11-q13.
Sintomi minori
- Riduzione dei movimenti fetali, pianto debole nel lattante progressivamente risolventesi
- Caratteristiche comportamentali: eccessi di collera, violente escandescenze e comportamento ossessivo-compulsivo
- Disturbi del sonno e apnee durante il sonno
- Bassa statura
- Ipopigmentazione
- Mani piccole (<25° cent.) e/o piedi piccoli (<10° cent.)
- Mani strette con margine ulnare rettilineo
- Anomalie oculari (esotropia, miopia)
- Saliva densa e vischiosa con croste agli angoli della bocca
- difetti nell'articolazione del linguaggio
- Lesioni cutanee da grattamento (skin picking)
Altri sintomi
- Elevata soglia del dolore
- Diminuito riflesso del vomito
- Alterazioni della termoregolazione nella 1ª infanzia o alterata sensibilità alla temperatura nella 2ª infanzia
- Scoliosi e/o cifosi
- Adrenarca precoce
- Osteoporosi
- Abilità nei giochi di pazienza (puzzle)
- Normali indagini neuromuscolari
Trattamento
Il trattamento abilitativo, per quanto concerne l'età pediatrica, si basa su sedute fisioterapiche (a fronte della condizione di ipotono frequente), logopediche (per affrontare il ritardo del linguaggio e le disprassie bucco-fonatorie), educative e psicomotorie (per far fronte alla condizione di ritardo mentale, acquisire quindi i pre-requisiti dell'apprendimento per l'ingresso alla scuola primaria e per gestire il discorso di ipotonia e disprassia presente per incoraggiare la coordinazione motoria del bambino).
Bibliografia
- Giovanni Neri, Maurizio Genuardi, Genetica umana e medica, Elsevier, 2010, ISBN 88-214-3172-X.
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su sindrome di Prader-Willi
Wikimedia Commons contiene immagini o altri file su sindrome di Prader-Willi
Collegamenti esterni
- (EN) Sindrome di Prader-Willi, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.
- Federazione Nazionale Sindrome di Prader Willi, su praderwilli.it.
| Controllo di autorità | Thesaurus BNCF 67274 · LCCN (EN) sh85106050 · GND (DE) 4201277-6 · BNF (FR) cb12468434q (data) · J9U (EN, HE) 987007531653205171 · NDL (EN, JA) 01179976 |
|---|