
| Distrofia miotonica | |
|---|---|
| Malattia rara | |
| Cod. esenz. SSN | RFG090 |
| Specialità | neurologia |
| Eziologia | mutazione |
| Classificazione e risorse esterne (EN) | |
| OMIM | 160900 |
| MeSH | D009223 |
| GeneReviews | Panoramica e Panoramica |
| Sinonimi | |
| Miotonia distrofica | |
| Eponimi | |
| Hans Gustav Wilhelm Steinert | |

La distrofia miotonica (DM) è una malattia genetica neuromuscolare degenerativa a carattere autosomico dominante, caratterizzata da un quadro clinico ampiamente variabile e da un decorso lentamente progressivo, il cui esordio può avvenire a qualunque età. Rappresenta la seconda forma di distrofia muscolare più diffusa dopo la distrofia muscolare di Duchenne.
Il quadro clinico è caratterizzato da perdita di massa muscolare, miotono, cataratta, difetti nel sistema di conduzione cardiaco, alterazioni endocrine e deficit cognitivi nei casi congeniti infantili di DM1. È presente un fenomeno di anticipazione per cui l'esordio tende a presentarsi ad un'età sempre più giovane di generazione in generazione nella stessa famiglia.
I pazienti con distrofia miotonica di tipo 1 o di Steinert, tranne che nella forma tardiva, hanno un'aspettativa di vita più bassa, essendo soggetti ad aritmia grave e cardiomiopatia (che possono esitare in morte cardiaca improvvisa), e a disturbi respiratori seri. Nella distrofia miotonica di tipo 2 o di Ricker l'aspettativa di vita del paziente può anche essere normale.
Epidemiologia
La prevalenza è stata calcolata intorno ad 1 su 8.000 persone.
Classificazione
Esistono varie forme di distrofia miotonica riconoscibili mediante l'esame del DNA:
- Distrofia miotonica di tipo 1 (DM1), detta anche malattia di Steinert, individuata nel 1909, che può avere un esordio precoce nei lattanti e bambini.
- Distrofia miotonica di tipo 2 (DM2) o PROMM (proximal myotonic myopathy), detta anche malattia di Ricker
Il 98% circa di tutti i casi di distrofia miotonica rientrano nel tipo 1, ad ogni modo il tipo 2 è caratterizzato da presentazione atipica e fenotipi insoliti, con sintomi diversi dalla forma classica, pertanto verosimilmente la diagnosi è sottostimata.
Gli specialisti stanno attualmente indagando sull'esistenza di altri tipi di distrofia miotonica.
Genetica
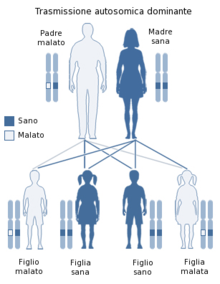
La Distrofia Miotonica è una malattia genetica a trasmissione autosomica dominante, pertanto si sviluppa in seguito all'acquisizione del gene affetto da un genitore. La probabilità di ereditare il gene affetto è del 50%.
La DM è caratterizzata dalla presenza di triplette nucleotidiche ripetute. Le ripetizioni di coppie o triplette nucleotidiche sono frequenti nel DNA, ma in questa patologia le ripetizioni sono in numero esagerato rispetto alle ripetizioni presenti nel DNA normale; questo fenomeno prende il nome di "amplificazione". Negli ultimi anni sono state identificate numerose malattie a trasmissione genetica in cui il meccanismo di danno è legato alla presenza di ripetizione di triplette, la prima ad essere studiata e la più nota fu la Corea di Huntington che condivide con la distrofia miotonica la caratteristica di esordire più precocemente ad ogni generazione successiva.
DM1
Nella forma DM1 (malattia di Steinert) il gene mutato è chiamato DMPK (myotonic dystrophy protein kinase) e codifica per una miosin chinasi espressa nei muscoli scheletrici. Questo gene è localizzato nel braccio lungo del cromosoma 19 (in posizione 19q13.3). In questo locus vi è un difetto molecolare specifico, costituito da una sequenza trinucleotidica instabile (Citosina Timina Guanina) nel 3'UTR che viene ripetuta 50-4000 volte quando nella popolazione normale il range varia da 5 a 37 volte.
DM2
Nella forma DM2 (detta anche miopatia miotonica prossimale/PROMM o malattia di Ricker) analogamente c'è un difetto nel gene ZNF9 sul cromosoma 3 in posizione 3q21. Il numero di ripetizioni va da 75 ad oltre 11000, ma in questo caso non sembra esserci differenza nella gravità della patologia o nella precocità di esordio. Il fenomeno dell'anticipazione in questa forma sembra essere meno significativo e in letteratura è riferita solo una lieve anticipazione. In questo caso la ripetizione coinvolge quattro nucleotidi.
Quadro clinico
Il quadro clinico è variabile, frequente nella DM1 il riscontro di miopatia, disartria, atrofia, ipotiroidismo, ritardo mentale (solo se esordio infantile), molto grave può essere il coinvolgimento del miocardio portando a forme di cardiomiopatia diffusa. La forma congenita e quella infantile sono gravi, mentre lo sono meno la forma giovanile-preadolescenziale e quella dell'adulto. La forma meno grave è la tardiva.
Nella DM2 si osservano frequentemente nistagmo,disfagia e dolore addominale mentre nella DM1 si osservano disturbi dello spettro autistico fra cui la sindrome di Tourette.
I pazienti con DM2 si presentano riferendo dolori muscolari, affaticabilità, rigidità, debolezza ai muscoli prossimali dell'arto inferiore (coscia). La forma meno grave è chiamata miopatia miotonica prossimale.
La DM presenta spesso anche problemi endocrini (ipotiroidismo e ipertiroidismo, diabete mellito), cataratta precoce, problemi gastrointestinali, ipersonnia, disturbi psichiatrici, problemi respiratori.
La distribuzione della debolezza muscolare è diversa per le due forme: nella DM1 sono affette le masse muscolari del volto e della mandibola, con ptosi palpebrale, debolezza dei muscoli del collo, delle mani e della parte distale della gamba (piede). Nella DM2 la debolezza è più evidente ai muscoli prossimali, quindi vicino al tronco: nuca, spalle, flessori dell'anca e parte superiore delle gambe.
Diagnosi
La diagnosi di distrofia miotonica può essere difficile poiché comporta la diagnosi differenziale con le patologie neuromuscolari che condividono alcuni aspetti del quadro clinico. Le patologie neuromuscolari sono perlopiù rare ed attualmente se ne conoscono oltre 40 che diventano 100 se si prendono in considerazione i sottotipi. Perciò un paziente con un quadro clinico complesso che potrebbe essere affetto da DM dovrà essere indirizzato ad uno specialista, ma a seconda del sintomo d'esordio il paziente potrebbe essere indirizzato ad un neurologo, ad un cardiologo, ad un oculista, ad un endocrinologo o ad un reumatologo. Poiché la presentazione è frequentemente atipica è possibile che se non viene interpellato un esperto di patologie neuromuscolari la diagnosi non venga eseguita.
Nonostante non esistano ad oggi cure specifiche per la DM e la terapia sia basata su interventi sintomatici (si affrontano i problemi mano a mano che si presentano), è importante che la diagnosi venga formulata correttamente, sia per monitorare il paziente e poter riconoscere precocemente le manifestazioni più gravi e potenzialmente fatali (ad esempio i problemi cardiaci), sia per fornire una consulenza genetica rispetto all'elevato rischio di trasmissione alla prole.
Il rischio anestesiologico è tale per cui la presenza di DM deve essere riferita ad ogni visita medica, anche per problemi non correlati alla patologia.
La diagnosi di certezza si ha mediante l'esame del DNA.
Terapia
Non esistono trattamenti efficaci contro tale forma di distrofia muscolare, per combattere in parte la miotonia si somministra la mexiletina (dosi 75 mg - 150 mg).
L'attenzione si concentra sulla gestione delle complicanze della malattia, in particolare quelle relative al sistema cardiopolmonare in quanto rappresentano il 70% dei decessi dovuti a DM1. L'inserimento di pacemaker può essere richiesto per soggetti con anomalie della conduzione cardiaca. Il miglioramento della qualità della vita che può essere misurato utilizzando questionari specifici è anche un obiettivo principale delle cure mediche. L'apnea centrale del sonno o l'apnea ostruttiva del sonno può causare eccessiva sonnolenza diurna e questi individui dovrebbero sottoporsi a uno studio del sonno. La ventilazione non invasiva può essere offerta in caso di anomalie respiratorie.
Alcuni piccoli studi hanno suggerito che l'imipramina, la clomipramina e la taurina possono essere utili nel trattamento della miotonia. Tuttavia, a causa della debole evidenza e dei potenziali effetti collaterali come aritmie cardiache, questi trattamenti sono usati raramente. Un recente studio del dicembre 2015 ha dimostrato che un comune antibiotico approvato dalla FDA, l'eritromicina ha ridotto la miotonia nei topi. Sono previsti studi sull'uomo per l'eritromicina. L'eritromicina è stata utilizzata con successo in pazienti con problemi gastrici.
È stato dimostrato che lo splicing alterato del canale 1 specifico per il muscolo (ClC-1) causa il fenotipo miotonico di DM1 ed è reversibile nei modelli murini che utilizzano l'antisenso morfolino per modificare lo splicing dell'mRNA di ClC-1.
Prognosi
La prognosi è infausta nella DM1 (tranne nella forma DM1 tardiva); il rischio di morte è più elevato nella tipologia 1 dato che è più soggetta ad aritmie fatali che causano morte improvvisa per torsione di punta. La DM2 è meno soggetta a morte cardiaca improvvisa della DM1 e alla cardiomiopatia.
La forma tardiva di DM1 è a volte asintomatica, mentre la forma miopatia miotonica prossimale di DM2 ha coinvolgimento cardiaco raro e prognosi migliore.
L'aspettativa di vita media di un paziente è generalmente più bassa, può essere però normale nelle due forme più lievi, specie nella DM2.
Bibliografia
- Joseph C. Segen, Concise Dictionary of Modern Medicine, New York, McGraw-Hill, 2006, ISBN 978-88-386-3917-3.
- Douglas M. Anderson, A. Elliot Michelle, Mosby’s medical, nursing, & Allied Health Dictionary sesta edizione, New York, Piccin, 2004, ISBN 88-299-1716-8.
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su distrofia miotonica
Wikimedia Commons contiene immagini o altri file su distrofia miotonica
Collegamenti esterni
- Searchable database Archiviato il 17 ottobre 2008 in Internet Archive. at Dutch Neuromuscular Research
- 140th ENMC International Workshop 2006 Myotonic Dystrophy DM2/PROMM and other Myotonic dystrophies]
- Disease Information from the Myotonic Dystrophy Foundation
- Information from the International Myotonic Dystrophy Organization
- MDSG Information, su mdsguk.org. URL consultato il 26 agosto 2009 (archiviato dall'url originale il 29 dicembre 2008).
- Information from the Neuromuscular Disease Center
- DM Toolbox Research tools for Myotonic Dystrophy from the Marigold Foundation
| Controllo di autorità | Thesaurus BNCF 43723 · LCCN (EN) sh85089286 · BNE (ES) XX4435101 (data) · BNF (FR) cb12268952p (data) · J9U (EN, HE) 987007555749205171 |
|---|