
| Malattia di Addison | |
|---|---|
| Specialità | endocrinologia |
| Classificazione e risorse esterne (EN) | |
| OMIM | 103230 |
| MeSH | D000224 |
| MedlinePlus | 000378 |
| eMedicine | 116467 |
| Eponimi | |
| Thomas Addison | |
La malattia di Addison (sovente chiamata anche morbo di Addison o insufficienza surrenalica cronica, ipocortisolismo, iposurrenalismo) è la forma primitiva di insufficienza corticosurrenale cronica che deriva da una severa riduzione, a carattere permanente e irreversibile, della increzione degli ormoni elaborati dal corticosurrene.
È dunque una malattia cronica del sistema endocrino caratterizzata da una serie di sintomi relativamente non specifici, come dolore addominale e debolezza, ma in determinate circostanze, questi possono evolversi con attacchi acuti gravi che possono comportare una severa ipotensione e il coma.
La condizione deriva da problemi alla ghiandola surrenale che possono essere dovuti a un malfunzionamento del sistema immunitario, da alcune infezioni o da varie cause più rare. La malattia di Addison è conosciuta anche come insufficienza corticosurrenale primaria cronica, per distinguerla dall'insufficienza corticosurrenale primaria acuta, spesso causata dalla sindrome di Waterhouse-Friderichsen.
La malattia di Addison dovrebbe essere distinta anche dall'insufficienza surrenalica secondaria e terziaria, che sono causate da mancanza di ACTH (prodotto dall'ipofisi) e CRH (prodotto dall'ipotalamo), rispettivamente. Nonostante questa distinzione, le crisi addisoniane possono accadere in tutte le forme di insufficienza surrenalica.
La malattia di Addison, e le altre forme di iposurrenalismo, sono generalmente diagnosticate tramite esami del sangue e tecniche di imaging medicale. Il trattamento consiste nel sostituire gli ormoni assenti (grazie a idrocortisone orale e fludrocortisone). È necessario mantenere la terapia sostitutiva steroidea in continuo, con un costante follow-up e il monitoraggio degli altri problemi di salute.
La condizione prende il nome da Thomas Addison, laureato presso l'Edinburgh Medical College, che per primo ne descrisse la condizione nel 1849. L'aggettivo "addisoniana" è usato per descrivere le caratteristiche della condizione, così come i pazienti affetti dalla malattia.
Storia

La malattia di Addison prende il nome da Thomas Addison, il medico britannico che per primo descrisse la patologia nella pubblicazione edita nel 1855 On the Constitutional and Local Effects of Disease of the Suprarenal Capsules . Originariamente la descrisse come "melasma suprarenale", ma in seguito la comunità scientifica le attribuì l'eponimo "malattia di Addison" in onore della scoperta da parte di Addison.
Tutti i sei pazienti iniziali di Addison presentavano tubercolosi delle ghiandole surrenali, mentre i sei relativi al 1855 mostravano sintomi di tubercolosi surrenalica; il termine "malattia di Addison" non implica un processo patologico sottostante.
La condizione era inizialmente considerata una forma di anemia associata alle ghiandole surrenali. Poiché all'epoca si sapeva poco delle ghiandole surrenali (allora chiamate "Capsule sopra-renali"), la monografia di Addison che descriveva la patologia fu un'intuizione isolata. Man mano che la funzione surrenale divenne maggiormente nota, lo studio di Addison divenne noto come un importante contributo scientifico e un classico esempio di attenta osservazione medica.
Epidemiologia
L'incidenza della malattia di Addison nella popolazione umana è stimata in circa 1 caso su 100 000 persone. Alcuni ricercatori, tuttavia, hanno proposto valori più elevati che vanno dai 40 ai 144 casi per milione di abitanti (rispettivamente, 1 su 25 000 e 1 su 7 000 abitanti). La patologia può colpire individui di qualsiasi età, sesso o etnia, ma in genere si presenta in adulti di età compresa tra 30 e 50 anni. Gli studi epidemiologici non hanno mostrato predisposizioni significative basate sull'etnia.
Eziologia
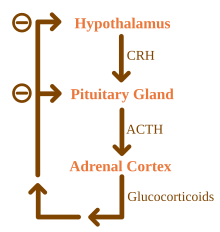
Le cause della malattia di Addison possono essere classificate in base al meccanismo che comporta un'insufficiente produzione di cortisolo da parte delle ghiandole surrenali. Può trattarsi di disgenesi surrenalica (la ghiandola non si è formata adeguatamente durante lo sviluppo), steroidogenesi compromessa (la ghiandola è presente ma è biochimicamente incapace di produrre cortisolo) o successiva a processi patologici che portano a una distruzione quasi completa della corteccia surrenalica.
Infine, la condizione può svilupparsi a seguito di un intervento di surrenectomia bilaterale in pazienti con forme particolarmente gravi di malattia di Cushing.
Distruzione del surrene
Circa il 70% dei casi di malattia di Addison è dovuto ad aggressione auto-immunitaria della ghiandola (atrofia surrenalica autoimmune o atrofia idiopatica della corteccia surrenale). Questa forma di malattia di Addison è caratterizzata da un infiltrato linfomonocitario (adrenalite) della corteccia surrenale. In circolo sono presenti ACA (Anti-Cortex Antibodies, anticorpi anti-corteccia) diretti verso la 21 idrossilasi, enzima chiave della steroidogenesi. Questi anticorpi sono molto specifici e occasionalmente si possono trovare anche in pazienti affetti da malattia di Basedow o tiroidite di Hashimoto e la loro presenza indica il rischio di sviluppo di insufficienza surrenalica. La rilevazione di questi anticorpi può precedere di anni l'insorgenza di un'insufficienza surrenalica manifesta. Il primo bersaglio sembra essere la zona glomerulare produttrice di aldosterone; infatti si osserva all'inizio un'elevazione dell'attività reninica del siero. Gli anticorpi, in seguito alla distruzione del surrene e quindi all'instaurarsi della sintomatologia clinica, scompaiono.
Nel mondo industrializzato, l'adrenalite autoimmune è la causa più comune della malattia di Addison. La distruzione autoimmune della corteccia surrenale è dovuta a una reazione immunitaria scatenata contro l'enzima steroide 21-monoossigenasi (un fenomeno descritto per la prima volta nel 1992). Questo enzima può essere isolato nel contesto della sindrome poliendocrina autoimmune (di tipo 1 o 2) in cui potrebbero essere coinvolti anche ulteriori organi deputati alla produzione di ormoni, come la tiroide e il pancreas.
La distruzione della ghiandola surrenalica è anche una caratteristica dell'adrenoleucodistrofia, della presenza di metastasi (diffusione di cellule tumorali provenienti altre parti del corpo, in particolare dal polmone) nelle ghiandole surrenali, del verificarsi di emorragie (ad esempio nella sindrome di Waterhouse-Friderichsen, infarti surrenali, o sindrome da anticorpi antifosfolipidi), nel caso di particolari infezioni (come tubercolosi, istoplasmosi, coccidiomicosi, AIDS) o con la deposizione di proteine anomale nei casi di amiloidosi.
Disgenesi surrenalica
Tutte le cause in questa categoria sono di origine genetiche e generalmente si riscontrano molto raramente. Tra queste vi possono essere le mutazioni del fattore di trascrizione SF1, l'ipoplasia surrenalica congenita (dovuta a mutazioni del gene DAX-1) e mutazioni al gene del recettore ACTH (o geni correlati, come nella sindrome della tripa A). Le mutazioni del gene DAX-1 possono raggrupparsi in una sindrome con deficit di glicerolo-chinasi che si associa a una serie di ulteriori altri sintomi, i quali compaiono quando tale gene viene eliminato insieme ad altri.
Fra le cause minori ricordiamo anche l'ipoplasia congenita dei surreni, malattia a carattere ereditario caratterizzata dalla mancata reattività delle cellule surrenali all'ACTH e malattia di Addison di origine iatrogena, da surrenectomia bilaterale, in pazienti con forme particolarmente gravi di malattia di Cushing.
Steroidogenesi compromessa
Per la sintesi del cortisolo la ghiandola surrenale ha bisogno di colesterolo, convertito poi biochimicamente in ormoni steroidei. Tra le patologie che comportano l'interruzione dell'approvvigionamento del colesterolo vi sono la sindrome di Smith-Lemli-Opitz e l'abetalipoproteinemia.
Tra i problemi di sintesi, l'iperplasia surrenalica congenita è la più comune (in varie forme: 21-idrossilasi, 17α-idrossilasi, 11β-idrossilasi e 3β-idrossisteride deidrogenasi), CAH lipoide a causa della carenza di mutazioni StAR e DNA mitocondriale. In rari casi la malattia di Addison può essere iatrogena, infatti alcuni farmaci interferiscono con gli enzimi di sintesi steroidea (ad esempio ketoconazolo), mentre altri accelerano la normale degradazione degli ormoni da parte del fegato (ad es. rifampicina, fenitoina).
Fisiopatologia
La carenza di aldosterone e cortisolo è responsabile delle manifestazioni più gravi della malattia di Addison, mentre la carenza di androgeni surrenalici è solo responsabile della riduzione dell'apparato pilifero nella donna, specie a livello ascellare.
La carenza di aldosterone determina una ridotta capacità di trattenere sodio (quindi anche di acqua) e di eliminare potassio a livello dei tubuli renali, perciò nella malattia di Addison si assiste all'aumento dell'escrezione di sodio e alla diminuzione dell'escrezione di potassio nelle urine che sono diluite: ne risultano basse concentrazioni ematiche di sodio e di cloro e un'alta concentrazione sierica di potassio. L'incapacità di concentrare le urine, associata alle modificazioni dell'equilibrio elettrolitico, determina una grave disidratazione, ipertonicità plasmatica, acidosi, ipovolemia, perdita di peso, ipotensione, diminuzione della gettata cardiaca, astenia intensa e facile insorgenza di episodi lipotimici da ipotensione ortostatica, inoltre l'iperkaliemia indotta da carenza di aldosterone può essere responsabile di disturbi del ritmo (asistolia, blocchi A-V, ecc.).
L'acido glicirretico possiede una molecola di struttura simile a quella dell'aldosterone (affinità bassa, ma definita, dunque che richiede alti dosaggi per avere un'attività biologica), un meccanismo d'azione e sintomatologia simili (tanto che l'ipokaliemia da liquirizia era una delle cause più frequenti di ipertensione nei Paesi dove la sua disponibilità non era limitata), un "agonismo" diretto acido glicirretico-aldosterone che permetteva a pazienti affetti dalla malattia di Addison di sopravvivere solo assumendo dosi elevate di radici di liquirizia: la carenza di aldosterone faceva aumentare il potassio e perdere acqua e sodio, mentre la liquirizia operava in modo contrario.
Il deficit di cortisolo contribuisce all'ipotensione e provoca disturbi metabolici come ridotta gliconeogenesi, diminuzione di glicogeno epatico, diminuita mobilizzazione e utilizzazione dei grassi, ipoglicemia che insieme all'iponatriemia sono responsabili dell'intensa astenia e della perdita di peso che caratterizza i pazienti addisoniani. Le alterazioni metaboliche vengono ritenute responsabili anche dei disturbi psichici che sono rilevabili clinicamente in circa 70% di questi pazienti e che consistono principalmente in apatia, ridotto interesse verso l'ambiente, depressione. Il cortisolo esercita normalmente un'azione stimolante l'eritropoiesi e influenza il traffico dei leucociti tra il compartimento intravascolare e i tessuti, inducendo aumento dei granulociti e diminuzione dei linfociti e degli eosinofili, la sua carenza determina modificazioni in senso opposto degli elementi ematici con anemia, neutropenia, linfocitosi ed eosinofilia. La riduzione dei livelli ematici di cortisolo comporta ipersecrezione di ACTH ipofisario e dei peptidi correlati come beta-lipotropina, alfa e beta-MSH, i quali hanno attività melanocito-stimolante e producono iperpigmentazione della cute e della mucosa caratteristica della malattia di Addison. La conseguenza più grave della mancanza di cortisolo è comunque rappresentata dall'incapacità dei pazienti addisoniani di rispondere adeguatamente a ogni tipo di stress fisiologico e patologico, questi soggetti risultano estremamente fragili di fronte a eventi morbosi, traumi, interventi chirurgici che sono agevolmente superati da soggetti normali.
Quadro clinico
Nella forma conclamata la malattia si esprime sintomatologicamente con una triade caratteristica: astenia, melanodermia, ipotensione. La progressione della malattia è lenta e graduale in rapporto alla progressione delle lesioni distruttive della ghiandola. Quando la perdita di tessuto surrenalico supera il 90% si ha un quadro completo di insufficienza surrenalica cronica. I principali sintomi e segni sono:
- astenia, ipoglicemia, affaticabilità e ipotensione ortostatica sono sintomi precoci, all'inizio l'astenia si manifesta dopo un periodo di stress, mentre successivamente il paziente è costretto a rimanere a letto. Al pari dell'astenia, anoressia e la perdita di peso sono elementi clinici costantemente presenti nell'Addison tanto che tale diagnosi in un paziente che goda di buon appetito o che presenti un incremento ponderale o che denunci un buon vigore fisico è improbabile.
- l'Acantosi nigricans e la melanodermia, vale a dire iperpigmentazione cutanea, costituiscono la seconda manifestazione cardinale della malattia e sono dovute a ipersecrezione di ACTH, ormone che deriva dallo stesso precursore dell'MSH (ormone che stimola i melanociti). Tale precursore è detto proopiomelanocortina (POMC). La iperpigmentazione nelle fasi iniziali è lieve e può essere confusa con residui di un'abbronzatura o con un naturale colorito bruno della pelle. Alcune caratteristiche dell'iperpigmentazione addisoniana permettono di distinguerla da altre forme: essa è più evidente a livello delle pieghe cutanee (specie alle pliche dei palmi delle mani), nelle zone esposte a pressione o a continui attriti (come nel solco sottomammario nelle donne che portano il reggiseno, o in corrispondenza del colletto, della cintura dei pantaloni, dei gomiti e delle ginocchia) e in tutte le regioni normalmente iperpigmentate (come areole mammarie, la regione scrotale e perineale) e ancora in sede di cicatrici prodotte successivamente all'esordio della malattia. Inoltre nella malattia di Addison la iperpigmentazione si estende anche alle mucose, fra queste quelle più frequentemente colpite sono la mucosa orale in corrispondenza delle guance, delle gengive, del palato, la mucosa vaginale e quella rettale. L'associazione fra melanodermia cutanea e iperpigmentazione mucosa è fortemente suggestiva per la natura addisoniana delle deviazioni pigmentarie. Va tuttavia aggiunto che fra le zone di iperpigmentazione cutanee possono essere intercalate delle chiazze di ipopigmentazione che possono giungere sino alla vitiligine. La cute è particolarmente secca per la disidratazione dovuta alla deplezione sodica.
- Spesso sono presenti vertigini e attacchi sincopali
- turbe neuropsichiche come irritabilità, ansia e apatia, nonché difficoltà di concentrazione
- dolori addominali, soprattutto in regione epigastrica con nausea, vomito e diarrea (diagnosi differenziale con ulcera peptica)
- alterazioni della funzione sessuale sono piuttosto frequenti, l'amenorrea, la più comune di esse, può essere la conseguenza della perdita di peso cui va incontro il paziente addisoniano o esprimere la concomitanza di una insufficienza gonadica primitiva a genesi autoimmune. La perdita dei peli pubici e ascellari nella donna esprime la ridotta secrezione degli androgeni surrenalici.
Crisi surrenalica
Con "crisi surrenalica" o "crisi addisoniana" si intende una serie di sintomi che indicano una grave insufficienza surrenalica. Questa può verificarsi a seguito di una malattia di Addison non diagnosticata in precedenza, di un processo patologico che colpisce improvvisamente la funzione surrenalica (ad esempio, un'emorragia surrenalica) o di un problema intercorrente (ad esempio un'infezione o un trauma) in qualcuno noto per avere la malattia di Addison. Si tratta di un'emergenza medica e di una situazione potenzialmente pericolosa per la vita che richiede un trattamento di emergenza immediato.
I sintomi caratteristici sono: dolore improvviso e penetrante alle gambe, alla parte bassa della schiena o all'addome, vomito e diarrea intensi con conseguente disidratazione, bassa pressione sanguigna, sincope (perdita di coscienza e capacità di stare in piedi), ipoglicemia (ridotto livello di glucosio nel sangue), psicosi, confusione mentale, letargia grave, iponatriemia (basso livello di sodio nel sangue), iperkaliemia (elevato livello di potassio nel sangue), ipercalcemia (elevato livello di calcio nel sangue), convulsioni, febbre.
Esami di laboratorio
Alcuni sono molto aspecifici, come anemia normocitica con linfocitosi ed eosinofilia, più significative sono alcune alterazioni ematochimiche come:
- iponatriemia (<130 mEq/L), deplezione di sodio significa deplezione idrica, la quale a sua volta comporta ipovolemia e ipotensione
- iperkaliemia (>5 mEq/L),responsabile di aritmie ipocinetiche, e acidosi metabolica
- ipoglicemia
- aumento dell'urea plasmatica e della creatininemia per un meccanismo prerenale che dipende dalla ipovolemia. La contrazione della volemia può essere tale da indurre una riduzione del filtrato glomerulare e quindi un aumento delle scorie azotate. Il quadro di iperazotemia prerenale regredisce non appena si provvede a reintegrare il patrimonio idrico dell'organismo.
- i livelli plasmatici di renina e ACTH sono aumentati.
N.B.: quando insufficienza corticosurrenalica è provocata da un'inadeguata produzione di ACTH ipofisaria, i livelli degli elettroliti sono nella norma, e manca la melanodermia (cosiddetto "Addison bianco").
- L'ECG può mostrare rallentamento generalizzato del ritmo, bassi voltaggi e allungamento degli intervalli PR e QT.
Diagnosi
La diagnosi viene sospettata sulla base dei sintomi e dei segni e confermata dai test di laboratorio. L'insufficienza corticosurrenalica può essere diagnosticata dimostrando l'incapacità ad aumentare i livelli plasmatici di cortisolo o l'escrezione urinaria di cortisolo libero dopo la somministrazione di ACTH.
Test di valutazione per l'insufficienza corticosurrenalica: il test si esegue iniettando 250 microgrammi di ACTH sintetico (Cortrosyn) EV. Prima dell'iniezione il cortisolo plasmatico normale è compreso fra 5 e 25 microgrammi/dL (fra 138 e 690 nmol/L) e raddoppia fra i 30 e i 90 minuti, con un minimo di 20 microgrammi/dL (552 nmol/L). I pazienti con malattia di Addison hanno valori bassi o normali che non subiscono incrementi.
Distinzione tra insufficienza corticosurrenalica primitiva e secondaria: la maggior parte dei casi di ipocorticosurrenalismo secondario è provocata dalla distruzione dell'ipofisi. La TC o la RMN della sella possono quindi essere utili per escludere la presenza di tumori o atrofia. Nei pazienti con una patologia primitiva del surrene, il livello plasmatico di ACTH è elevato (> 50 pg/mL). I pazienti con insufficienza ipofisaria o con deficit isolato di ACTH hanno un basso livello dell'ormone. Se si sospetta un'insufficienza surrenalica secondaria, questa potrà essere confermata da test al metirapone. Il metirapone è un farmaco in grado di bloccare l'enzima 11-b-idrossilasi, quindi, deprime la conversione dell'11-desossicortisolo in cortisolo, i cui livelli plasmatici si riducono con conseguente incremento della secrezione di ACTH.
Quest'ultimo stimola la steroidogenesi surrenalica, il che causa un accumulo di 11-b-idrossicortisolo, lo steroide che immediatamente precede la tappa enzimatacamente bloccata. Poiché l'11-desossicortisolo viene metabolizzato ed escreto in forma di 17-OHCS, l'eliminazione urinaria di questi steroidi risulterà notevolmente potenziata dalla somministrazione di metirapone. Il metodo migliore e più semplice è quello di somministrare a mezzanotte 30 mg/kg di metirapone PO insieme con una piccola quantità di cibo per evitare l'irritazione gastrica. Il cortisolo plasmatico alle 8 del mattino seguente deve essere < 10 microgrammi /dl (< 276 nmol/L) e l'11-deossicortisolo plasmatico deve essere tra 7 e 22 microgrammi/dL (tra 0,2 e 0,6 mmol/L). Nei pazienti che non rispondono al metirapone, deve essere eseguito un test all'ACTH. I pazienti con insufficienza corticosurrenalica primitiva hanno bassi livelli di entrambi gli steroidi e non rispondono all'ACTH; quelli con ipopituitarismo rispondono all'ACTH, ma non al metirapone.
Iposurrenalismo acuto o crisi surrenalica acuta è una condizione di emergenza medica caratterizzata dalla brusca insorgenza di shock rapidamente ingravescente, questa può insorgere in seguito a errori dietetici (alimenti ricchi di potassio, diete povere di sale) o più spesso a causa dell'interruzione o del ritardo di inizio della terapia corticosteroidea sostitutiva o infine nel corso di un evento stressante, come un'infezione, un trauma, un intervento chirurgico che colpisca un individuo affetto da una insufficienza corticosurrenalica misconosciuta e fino a quel momento in equilibrio labile o in un paziente in terapia sostitutiva, in cui non si sia provveduto a un opportuno aumento della posologia di corticosteroidi imposta dal sopraggiungere dello stress.
Le forme più gravi di insufficienza corticosurrenalica acuta conseguono alla brusca, completa e irreversibile distruzione della ghiandola causata da emorragie massicce a livello surrenale in corso di sepsi, di leucemie acute, di terapia anticoagulante massiva, di trombosi della vena centrale. La sepsi meningococcica (sindrome di Waterhouse-Friderichsen) è nel bambino la causa più frequentemente responsabile di insufficienza surrenalica acuta. Il quadro clinico insorge acutamente, spesso in forma esplosiva, ed è caratterizzato da: profonda astenia, dolori addominali violenti a sede epigastrica, accompagnati frequentemente da vomito e diarrea profusa (al punto da fare sospettare una erronea diagnosi di gastroenterite acuta o suggerire l'ipotesi di un addome acuto), collasso vascolare periferico e, infine, insufficienza renale acuta con iperazotemia. La temperatura corporea può essere al di sotto della norma benché, spesso, si osservi grave ipertermia dovuta alle infezioni. Nella sindrome di Waterhouse-Friderichsen, l'infezione si accompagna a manifestazioni emorragiche diffuse che, oltre al surrene, interessano la cute (purpura fulminans), l'encefalo, i visceri.
Alcuni elementi vengono comunemente indicati come distintivi dello shock iposurrenalico: spiccata disidratazione, tendenza all'ipoglicemia, iperkaliemia e iposodiemia, estrema riduzione del tasso del cortisolo ematico. Nella pratica questi dati hanno limitato valore diagnostico in quanto l'evoluzione dell'iposurrenalismo acuto è così rapida e tumultuosa da non consentire lo svolgimento delle principali tappe dell'iter diagnostico. La diagnosi è pertanto fondamentalmente clinica. La possibilità di un'insufficienza surrenalica acuta deve essere presa in considerazione in ogni paziente in collasso, e se qualche elemento anamnestico e obiettivo anche vago avvalora il nostro sospetto (pregressa terapia steroidea, insorgenza nel decorso di una sepsi o dopo gravi traumi addominali o lombari) è bene incominciare immediatamente la terapia, somministrando dosi massicce di corticosteroidi e reintegrando la perdita di sodio con infusioni di NaCl isotoniche.
Trattamento
Il trattamento è sostitutivo, vale a dire va fatto con le sostanze (ormoni) che la ghiandola non produce o produce in quantità insufficiente. Questo vale naturalmente per tutti i deficit funzionali ghiandolari (vedi ad esempio le isole di Langherans e la ridotta o nulla produzione di insulina).
Prognosi
Generalmente, la prognosi della malattia di Addison è buona, se tale condizione viene correttamente trattata. La maggior parte dei pazienti può aspettarsi di vivere una vita relativamente normale. Chi soffre della malattia dovrebbe prestare attenzione al presentarsi di sintomi di una "crisi di Addison" quando si trova in un momento di stress, come durante un esercizio fisico, in cui necessiterebbe di un trattamento di emergenza con iniezioni endovenose per gestire la situazione.
Gli individui con la malattia di Addison presentano un tasso di mortalità più che raddoppiato. Inoltre, coloro che hanno anche il diabete mellito presentano un aumento della mortalità di quasi 4 volte rispetto alle persone con solo diabete.
Negli altri animali
Si riscontrano casi della malattia anche nei cani: è una forma estremamente rara che colpisce 1 cane su 10 milioni.
Tuttavia, nei cani è particolarmente difficile da diagnosticare, dando sintomi quali astenia o letargia, che possono essere confusi con altre patologie, quindi l'incidenza può essere più alta. Si nota inoltre una lieve prevalenza dell'insorgenza in alcune razze e nella fascia di età dai sei anni.
Bibliografia
- (EN) Robert Volpé, Autoimmune Diseases of the Endocrine System, CRC Press, 1990, ISBN 978-0-8493-6849-3.
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su malattia di Addison
Wikimedia Commons contiene immagini o altri file su malattia di Addison
Collegamenti esterni
- Addison, morbo di-, su sapere.it, De Agostini.
- (EN) Malattia di Addison, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.
- Sito dell'Associazione di Pazienti di Addison AIPAd, su morbodiaddison.org.
|
Classificazione e risorse esterne (EN) |
ICD-9-CM: 255.41; ICD-10-CM: E27.1; OMIM: 103230; MeSH: D000224; DiseasesDB: 222; |
| Controllo di autorità | Thesaurus BNCF 21285 · LCCN (EN) sh85000815 · BNE (ES) XX529072 (data) · BNF (FR) cb124303775 (data) · J9U (EN, HE) 987007292953405171 |
|---|