
| Malattia di Creutzfeldt-Jakob | |
|---|---|
 | |
| Specialità | neurologia e infettivologia |
| Eziologia | prione |
| Classificazione e risorse esterne (EN) | |
| OMIM | 123400 |
| MeSH | D007562 |
| MedlinePlus | 000788 |
| eMedicine | 1169688 |
| Eponimi | |
| Hans Gerhard Creutzfeldt Alfons Maria Jakob | |
La malattia di Creutzfeldt-Jakob (MCJ), originariamente descritta negli anni venti del XX secolo da Hans Gerhard Creutzfeldt ed Alfons Maria Jakob, è una malattia neurodegenerativa rara, che conduce ad una forma di demenza progressiva fatale.
La sindrome clinica è caratterizzata da deficit polisettoriali prevalentemente corticali con perdita di memoria, cambiamenti di personalità, allucinazioni, disartria, mioclono, rigidità posturale e convulsioni. La malattia di Creutzfeldt-Jakob è la forma più frequente di encefalopatia spongiforme umana. A livello istologico si assiste alla formazione di microvacuolazioni del tessuto cerebrale che assume una struttura di tipo spugnoso, dovute alla progressiva perdita di neuroni causata da alterazione di una proteina di membrana, espressa prevalentemente in cellule del sistema nervoso e del sistema reticolo-endoteliale, il prione. La malattia di Creutzfeldt-Jakob appartiene al gruppo delle encefalopatie spongiformi trasmissibili, una tipologia di malattie neurodegenerative dovute alla presenza di prioni. Il prione nella sua forma alterata (PrPSc) dimostra capacità infettive, potendo cioè propagarsi agendo sulla forma nativa (PrPc), facendo rientrare la malattia di Creutzfeldt-Jakob tra le encefalopatie trasmissibili, anche se non si considera contagiosa in senso tradizionale.
L'incidenza della MCJ si è mantenuta relativamente costante negli ultimi 80 anni, nell'ordine di 1-2/1.000.000/anno. Ottiene gli onori della cronaca dopo la descrizione dei primi casi di una forma variante, ancor più rara, legata all'epidemia di encefalopatia spongiforme bovina, la cosiddetta malattia della "mucca pazza".
Storia
La malattia è stata descritta per la prima volta nel 1920 dal neurologo tedesco Hans Gerhard Creutzfeldt e poco dopo da Alfons Maria Jakob, da cui il nome di Creutzfeldt-Jakob. Alcuni dei risultati clinici descritti nei loro primi lavori non corrispondono ai criteri attuali per la malattia, ed è stato ipotizzato che almeno due dei pazienti osservati negli studi iniziali siano in realtà stati affetti da una malattia diversa. Una prima descrizione della forma familiare si ha dal neurologo e psichiatra tedesco Friedrich Meggendorfer (1880-1953).
Epidemiologia

Anche se MCJ era la più comune malattia umana causata da prioni, la sua presentazione è ancora rara, e si verifica in circa un caso su un milione di persone ogni anno. Solitamente colpisce persone di età tra i 45 e i 75 anni e appare più comunemente nelle persone di età compresa tra i 60 e i 65. L'eccezione a ciò è la più recente variante (vMCJ), che si verifica nelle persone più giovani.
Il Centers for Disease Control and Prevention (CDC) degli Stati Uniti monitora la presenza della malattia nel paese, attraverso revisioni periodiche dei dati nazionali di mortalità. Secondo il CDC:
- La MCJ si verificava in tutto il mondo ad una frequenza di circa 1 caso ogni milione di abitanti all'anno.
- Sulla base della sorveglianza della mortalità rilevata tra il 1979 e il 1994, l'incidenza annuale di MCJ era rimasta stabile a circa 1 caso ogni milione di persone negli Stati Uniti.
- Negli Stati Uniti, i morti per MCJ tra le persone di età inferiore ai 30 anni di età si verificavano in una bassissima percentuale.
- La malattia si trovava frequentemente in pazienti con tra i 55 e i 65 anni di età, ma in alcuni casi poteva in persone di età superiore ai 90 anni e più giovani dei 55 anni di età.
- In più dell'85% dei casi, la durata della malattia, dopo l'insorgenza dei sintomi, era inferiore ad un anno (mediana: quattro mesi), portando a morte certa.
Per quanto riguarda la nuova variante, dal 1996 a marzo 2014 il numero totale di casi si assestò a 225, di cui 177 nella sola Gran Bretagna. Il grafico a destra ritraeva le nazioni colpite dall'epidemia della forma variante e del suo corrispettivo bovino.
Classificazione
I tipi di MCJ includono:
- Forma sporadica (sMCJ)
- Forma familiare (fMCJ)
- Forma iatrogena (iMCJ)
- Nuova variante (vMCJ) Identificata per la prima volta nel 1996.
Segni e sintomi
Il primo sintomo della MCJ è una veloce e progressiva demenza che porta alla perdita di memoria, a cambiamenti di personalità e allucinazioni. Questo è accompagnato da problemi fisici come disturbi del linguaggio, rapidi movimenti involontari (mioclono), disfunzioni dell'equilibrio e della coordinazione (atassia), cambiamenti nella marcia, postura rigida e convulsioni. La durata della malattia varia notevolmente, portando a morte in molti mesi o poche settimane (Johnson, 1998). In alcune persone, i sintomi possono continuare per anni. Nella maggior parte dei pazienti, questi sintomi sono seguiti da movimenti involontari e dalla comparsa di un elettroencefalogramma atipico. La maggior parte dei pazienti muore a sei mesi dall'esordio, spesso a causa di infezioni intercorrenti quali polmoniti dovute al deterioramento del riflesso della tosse. Circa il 15% dei pazienti sopravvive per due o più anni.
I sintomi di MCJ sono causati dalla progressiva morte delle cellule nervose del cervello, che è associata con la formazione di placche amiloidi dovuti ai prioni. Quando il tessuto cerebrale di un paziente affetto dal malattia di Creutzfeldt-Jakob viene esaminato al microscopio, molti piccoli fori possono essere visti, dove intere aree delle cellule nervose sono morte. La parola "spongiforme" in "encefalopatie spongiforme" si riferisce all'aspetto spugnoso del tessuto cerebrale.
Eziopatogenesi
Le encefalopatie spongiformi trasmissibili sono malattie causate da prioni. Le malattie sono così a volte chiamate "malattie da prioni". Tra le altre malattie da prioni che colpiscono gli esseri umani vi è la malattia di Gerstmann-Sträussler-Scheinker (GSS), insonnia familiare fatale (IFF) e la kuru, così come l'encefalopatia spongiforme bovina (BSE, comunemente conosciuta come morbo della mucca pazza) nei bovini, la malattia del deperimento cronico del cervo (CWD), e la scrapie nelle pecore. Anche la malattia di Alpers dei neonati è ritenuta essere una encefalopatia spongiforme trasmissibile causata da un prione.
Il prione ritenuto essere la causa della Creutzfeldt-Jakob presenta almeno due conformazioni stabili. La prima, allo stato nativo, è solubile in acqua ed è presente nelle cellule sane. A partire dal 2007 si ritiene che la sua funzione biologica sia presumibilmente implicata nel trasporto transmembrana o nella segnalazione. L'altro stato conformazionale è relativamente insolubile in acqua e forma facilmente aggregati proteici.
Le persone possono anche acquisire la malattia geneticamente attraverso una mutazione del gene che codifica per la proteina prionica (PRNP). Ciò però si verifica in solo 5-10% di tutti i casi di malattia di Creutzfeldt-Jakob.
Il prione della Creutzfeldt-Jakob è pericoloso perché, durante la malattia, favorisce il ripiegamento scorretto delle proteine. Il numero di molecole proteiche mal ripiegate aumenta esponenzialmente e il processo forma una grande quantità di proteine insolubili nelle cellule colpite. Questa massa di proteine mal ripiegate distrugge la funzionalità delle cellule e provoca la morte cellulare. Una volta che il prione viene trasmesso, le proteine difettose invadono il cervello.
A Stanley B. Prusiner dell'Università della California, San Francisco (UCSF) è stato assegnato il Premio Nobel per la fisiologia e per la medicina nel 1997 per la sua scoperta dei prioni. Per più di un decennio, la neuropatologa della Yale University Laura Manuelidis si è impegnata nella difficile spiegazione per la malattia. Nel gennaio 2007, lei e i suoi colleghi hanno pubblicato un articolo dove affermavano di aver trovato delle particelle simili ai virus (ma senza trovarvi gli acidi nucleici) in meno del 10% delle cellule di topo infettate dall'agente umano della malattia di Creutzfeldt-Jakob.
Trasmissione
La proteina difettosa può essere trasmessa da: innesti corneali, innesti durali o tramite impianti di elettrodi (forma acquisita o iatrogena: iMCJ), ma essa può anche essere ereditata (forma ereditaria o familiare: fMCJ), oppure può comparire per la prima volta nel paziente (forma sporadica: MCJ). Nella forma ereditaria, si verifica una mutazione nel gene per PrP, il PRNP. Dal 10 al 15% dei casi di MCJ sono ereditari.
La malattia inoltre è stato indicata come il risultato di un uso dell'ormone umano della crescita ottenuto dalle ghiandole pituitarie di persone che sono morte di malattia di Creutzfeldt-Jakob, anche se l'incidenza conosciuta per questa causa è (a partire da aprile 2004) piuttosto piccola. Negli Stati Uniti il farmaco che utilizza l'ormone da cadavere è stato ritirato nel 1985, mettendo fine a questo tipo di trasmissione nel paese.
Si ritiene che l'uomo possa contrarre la malattia consumando carni di animali infettati con la forma bovina della malattia.
Il cannibalismo è stato anche coinvolto come una causa di trasmissione per i prioni anomali, causando una malattia nota come kuru, riscontrata soprattutto tra le donne e i bambini della popolazione Fore in Papua Nuova Guinea. Mentre gli uomini della tribù mangiano il corpo del defunto e raramente contraggono la malattia, le donne e i bambini, che mangiavano le parti del corpo meno desiderabili, compreso il cervello, erano 8 volte più propense degli uomini a contrarre la kuru dal tessuto infetto.
I prioni, l'agente infettivo della MCJ, non possono essere inattivati mediante procedure di sterilizzazione post-chirurgica. L'Organizzazione Mondiale della Sanità e il Centers for Disease Control and Prevention raccomandano che lo strumentario utilizzato in questi casi debba essere immediatamente distrutto dopo l'uso. Tuttavia, nessun caso di trasmissione iatrogena di MCJ è stato riportato successivamente alla adozione delle attuali procedure di sterilizzazione e comunque nessuna a partire dal 1979.
Restrizioni per i donatori di sangue
Nel 2004 un nuovo rapporto pubblicato sulla rivista medica The Lancet ha dimostrato che la vMCJ può essere trasmessa attraverso trasfusioni di sangue. La scoperta ha allarmato i funzionari sanitari poiché ciò potrebbe portare ad una vasta epidemia nel futuro. Non esiste un test per determinare se un donatore di sangue è stato infettato con vMCJ. In reazione a questa relazione, il governo britannico ha impedito a chiunque avesse ricevuto una trasfusione di sangue, a partire dal gennaio 1980, la donazione di sangue. A partire dal 1999 è stato posto il divieto in Gran Bretagna per l'utilizzo del sangue del Regno Unito per la realizzazione di prodotti frazionari come l'albumina.
Il 28 maggio 2002, la Food and Drug Administration istituì una regola che esclude dalla donazione chiunque abbia trascorso almeno sei mesi in alcuni paesi europei (o tre mesi nel Regno Unito), dal 1980 al 1996. Dato il gran numero di militari statunitensi e ai loro familiari che risiedono in Europa, ci si aspettava che oltre il 7% dei donatori sarebbe stato fermato a causa della politica. In seguito le modifiche di questa politica hanno allentato la restrizione per un totale cumulativo di cinque anni o più da un viaggio civile nei paesi europei (sei mesi o più, se militare). I tre mesi di restrizione sui viaggi nel Regno Unito, tuttavia, non sono stati modificati.
A Singapore la Croce Rossa esclude potenziali donatori che hanno trascorso un periodo complessivo di tre mesi o più nel Regno Unito tra il 1980 e il 1996.
In Nuova Zelanda, chi ha vissuto nel Regno Unito, in Francia o nella Repubblica d'Irlanda per un totale di sei mesi o più tra gennaio 1980 e dicembre 1996 è stato escluso definitivamente dalla donazione di sangue. Chiunque abbia ricevuto una trasfusione di sangue in quei paesi a partire dal gennaio 1980 è anche precluso in modo permanente.
Regolamenti analoghi sono in atto anche in Germania e in Italia, dove chi ha passato sei mesi o più di vita nel Regno Unito tra gennaio 1980 e dicembre 1996 viene permanentemente escluso dalla donazione di sangue.
In Polonia, chiunque abbia trascorso cumulativamente sei o più mesi tra il 1º gennaio 1980 e il 31 dicembre 1996 nel Regno Unito, Irlanda o Francia è definitivamente escluso dalla donazione.
In Svizzera la possibilità di donare il sangue viene preclusa in modo permanente a coloro i quali abbiano ricevuto una trasfusione di sangue intero a partire dal 1º gennaio 1980 e se hanno vissuto nel Regno Unito, e quindi Inghilterra, Galles, Scozia, Irlanda del Nord, Isola di Man, Isole del Canale, Isole Falkland e Gibilterra per più di sei mesi tra il 1980 e il 1996.
Diagnosi

Si sospetta una diagnosi di MCJ quando vi si riscontrano i tipici sintomi e segni clinici, come una demenza rapidamente progressiva con mioclono. Ulteriori analisi possono supportare tale sospetto diagnostico, tra cui:
- Elettroencefalografia, presenta talora caratteristici picchi trifasici
- Analisi del liquido cerebrospinale per la ricerca della proteina 14-3-3, e per la determinazione dei livelli di tau totale e fosforilata
- Risonanza magnetica dell'encefalo, mostra spesso elevata intensità di segnale nel nucleo caudato e putamen bilateralmente, o a livello corticale nelle immagini pesate in DWI (Diffusion-Weighted Imaging) o con metodica FLAIR (Fluid Attenuated Inversion Recovery).
- Uno studio del 2010 e del 2011 ha identificato un possibile test del sangue per la MCJ. Il test tenta di identificare il prione responsabile della malattia.
La metodica DWI offre le immagini più sensibili per il sospetto di CJD, con circa 80% dei pazienti che soddisfano i criteri radiologici. In circa il 24% dei casi è identificabile una iperintensità solo a livello corticale (il cosiddetto segno "a nastro corticale" o cortical ribboning, per il coinvolgimento elettivo della sostanza grigia), nel 68% anomalie corticali e dei nuclei della base e nel 5%, anomalie dei soli nuclei della base. Il coinvolgimento del talamo può essere trovato nella MCJ e in particolare nella vMCJ.
Il quadro clinico per MCJ è variegato e non consente una diagnosi univoca, la quale necessita anche di criteri strumentali o liquorali. I criteri più recenti stilati dal MRI-CJD Consortium criteria for sporadic Creutzfeldt–Jakob disease prevedono quanto segue
- Segni clinici
- Demenza
- Segni cerebellari o visivi
- Segni piramidali o extrapiramidali
- Mutismo acinetico
- Esami strumentali
MCJ Probabile: due item da 1. e almeno uno da 2
MCJ Possibile: due item da 1. e durata di malattia dall'esordio al decesso < 2 anni
Negli ultimi anni, vari studi hanno dimostrato che il marcatore tumorale neurone-specifico (Enolasi 2) è spesso elevato nei casi di MCJ, tuttavia la sua utilità diagnostica è vista soprattutto quando viene associato ad un test per la proteina 14-3-3. Al 2012, i test di screening per identificare gli individui infetti asintomatici, da utilizzarsi ad esempio per i donatori di sangue, non sono ancora disponibili, anche se vari metodi sono stati proposti e valutati.

Nel 2010, un gruppo di ricercatori di New York individua il PrPSc anche se inizialmente presente in una sola parte su 100 miliardi (10−11) di tessuto cerebrale. Il metodo combina l'amplificazione con una nuova tecnologia chiamata Surround Optical Fiber Immunoassay (SOFIA) e alcuni anticorpi specifici contro PrPSc. Dopo l'amplificazione vengono concentrati i PrPSc, i campioni sono quindi etichettati con un colorante fluorescente utilizzando un anticorpo specifico e infine caricati in un micro-capillare. Questo tubo è collocato in un apparecchio costruito appositamente in modo che sia completamente circondato da fibre ottiche per catturare tutta la luce emessa quando il colorante viene eccitato con un laser.
I ricercatori hanno anche testato il loro metodo su campioni di sangue di pecora apparentemente sani che hanno continuato a sviluppare scrapie. Il cervello degli animali è stato analizzato quando tutti i sintomi erano divenuti evidenti. I risultati hanno mostrato chiaramente che il PrPSc poteva essere rilevato nel sangue degli animali prima che i sintomi apparissero. Dopo un ulteriore sviluppo e testing di questo metodo, potrebbe diventare di grande utilità come test di screening per la MCJ.
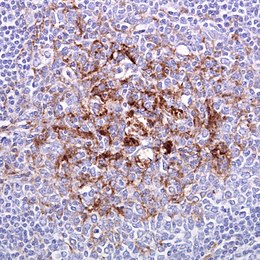
La diagnosi di vMCJ può essere supportato da una biopsia delle tonsille in caso di sospetta vMCJ, in quanto può contenere una quantità significativa di PrPSc. La biopsia del tessuto cerebrale è il test diagnostico definitivo in tutte le altre forme di MCJ e in caso di negatività alla biopsia tonsillare anche nella vMCJ. A causa della sua invasività, la biopsia non è effettuata se sulla base degli accertamenti precedenti il sospetto clinico è molto alto o troppo basso. Una biopsia negativa non esclude MCJ, dal momento che può essere predominante in una parte specifica del cervello.
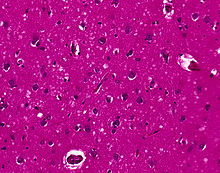
La materia grigia cerebrale assume il classico aspetto istologico spongiforme: la presenza di numerosi vacuoli tondi, dall'aspetto vitreo o eosinofilo, con la tendenza a confluire e di dimensione variabile tra 1 e 50 micrometri, localizzati in tutti i sei strati corticali nella corteccia cerebrale o con interessamento dello strato cerebellare. Possono essere anche notate perdita neuronale e gliosi. Placche di sostanza amiloide possono essere riscontrate nella neocorteccia nei casi di nuova variante della MCJ.
Purtroppo, la vacuolizzazione è presente anche in altri stati patologici. Diffuse vacuolizzazione corticali si notano anche nella malattia di Alzheimer e la vacuolizzazione superficiale corticale si verifica nelle ischemie e nella demenza frontotemporale. Questi vacuoli appaiono chiari. Vacuoli più grandi, che circondano i neuroni, i vasi e le glia, sono un possibile artefatto di elaborazione.
- Caratteristiche cliniche e patologiche:
| Caratteristica | MCJ classica | MCJ variante |
|---|---|---|
| Età media al decesso | 68 anni | 28 anni |
| Durata media della malattia | 4–5 mesi | 13–14 mesi |
| Segni clinici e sintomi | Demenza; primi segni neurologici | Sintomi psichiatrici/comportamentali; disestesie dolorose; segni neurologici ritardati |
| Onde aguzze periodiche su elettroencefalogramma | Talora presente | Spesso assente |
| Iperintensità di segnale nel nucleo caudato e nel putamen o a livello del nastro corticale in sequenze DWI e/o FLAIR | Spesso presente | Spesso presente |
| Analisi immunoistochimica del tessuto cerebrale | Accumuli variabili | Contrassegnato accumulo di proteina prionica resistente alle proteasi |
| Presenza dell'agente nel tessuto linfonodale | Non facilmente rilevato | Facilmente rilevato |
| Presenza di placche amiloidali nel tessuto nervoso | Possono essere presenti | Possono essere presenti |
- Un segnale anomalo nella parte posteriore del talamo nelle immagini T2 pesate, DWI e FLAIR di risonanza magnetica del cervello (MRI), in un contesto clinico appropriato, è altamente specifico per vMCJ. (Fonte: CDC)
Trattamento
Non è stato dimostrato alcun trattamento efficace per la MCJ, la malattia è sempre fatale e la ricerca per una cura continua. Un trattamento sperimentale è stato provato su un adolescente nordirlandese, Jonathan Simms, nel gennaio 2003. Il farmaco, chiamato pentosano polisolfoestere (PPS) viene generalmente utilizzato per trattare la cistite interstiziale e viene infuso nel ventricolo laterale del cervello del paziente. Il farmaco non sembra in grado di fermare la malattia e sia la funzione del cervello che dei tessuti continua ad essere compromessa. Tuttavia, il trattamento è ritenuto in grado di rallentare la progressione della malattia altrimenti incurabile e può aver contribuito alla più lunga sopravvivenza, rispetto alle attese, dei sette pazienti che sono stati studiati. Gli esperti del Dipartimento della Salute del Regno Unito non ritengono però i dati sufficienti per valutare l'utilizzo del pentosano polifosfato e raccomanda ulteriori ricerche su cavie animali. Nel 2007, la revisione del trattamento dei 26 pazienti con PPS non ha trovato alcuna prova di efficacia.
Gli scienziati hanno sperimentato l'utilizzo del RNA interference per rallentare la progressione della scrapie nei topi. Questa ricerca è improbabile che possa portare ad una terapia umana per molti anni.
Sia l'amfotericina B e che la doxorubicina sono state studiate come potenzialmente efficaci contro MCJ, ma ancora non vi è una forte evidenza che entrambi i farmaci siano efficaci nel fermare la malattia. Un ulteriore studio è stato provato con altri farmaci, ma nessuno è risultato efficace. Tuttavia, farmaci per ridurre la sofferenza esistono, e comprendono Valproato, un anticonvulsivante, e il clonazepam, per ridurre spasmi muscolari
Prognosi
Non essendoci una cura definitiva, la prognosi è infausta nella totalità dei casi. L'85% muore entro due anni, il restante 15% sopravvive più a lungo.
Aspetti particolari
In Italia è ben noto l'embargo di prodotti inglesi a base di carne bovina che è durato circa 10 anni, dal 1996 al 2006, in seguito alla forte epidemia di CJD bovina nel Regno Unito nel 1995. Per scongiurare la possibilità di trasmissione di CJD, le direttive del Ministero della Sanità si sono mosse anche nel campo della Medicina Trasfusionale, ribadendo a più riprese l'obbligo di escludere dalla donazione di plasma e midollo osseo coloro i quali avessero ricevuto impianti di cornea o chi avesse avuto in famiglia casi di SJC o insonnia familiare fatale.
Voci correlate
- Encefalopatia spongiforme bovina
- Encefalopatia spongiforme trasmissibile
- Kuru (malattia)
- Nuova variante di MCJ
- Prione
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su malattia di Creutzfeldt-Jakob
Wikimedia Commons contiene immagini o altri file su malattia di Creutzfeldt-Jakob
Collegamenti esterni
- (EN) Malattia di Creutzfeldt-Jakob, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.
- Associazione Italiana Encefalopatie da Prioni - AIEnP onlus, su aienp.it.
| Controllo di autorità | Thesaurus BNCF 54342 · LCCN (EN) sh85069294 · GND (DE) 4220958-4 · BNF (FR) cb12064993z (data) · J9U (EN, HE) 987007529248005171 · NDL (EN, JA) 00575235 |
|---|