
| Sindrome di Behçet | |
|---|---|
 | |
| Malattia rara | |
| Cod. esenz. SSN | RC0210 |
| Specialità | immunologia e reumatologia |
| Eziologia | sistema immunitario |
| Classificazione e risorse esterne (EN) | |
| OMIM | 109650 |
| MeSH | D001528 |
| eMedicine | 1006358, 1122381 e 1229174 |
| Eponimi | |
| Hulusi Behçet | |
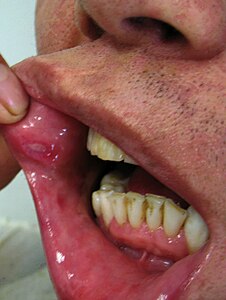
La sindrome di Behçet (pronuncia turca: [bɛˈtʃɛt]) è una malattia rara di origine autoimmune. Prende il nome dal dermatologo turco Hulusi Behçet, che per primo descrisse nel 1937 i tre principali sintomi che la contraddistinguono (ricorrenti afte orali e genitali oltre a uveite).
Si tratta di un disordine infiammatorio multisistemico recidivante caratterizzato da afte orali, genitali, uveite, tromboflebite e che frequentemente coinvolge le articolazioni, la cute, il sistema nervoso centrale e il tratto gastrointestinale. La sua eziologia è sconosciuta, benché alcuni studi scientifici abbiano ipotizzato un'associazione genetica con l'antigene HLA-B51 (e con l'HLA-B57 limitatamente alla popolazione caucasica).
È una malattia cronica che colpisce prevalentemente tra i 20 e i 30 anni; in Italia l'incidenza è di 2,4 casi ogni milione di abitanti e la prevalenza di 3,8/100 000. Dal punto di vista anatomopatologico si osservano lesioni importanti, anche se non specifiche, a carico dei piccoli vasi: le pareti di questi ultimi risultano infiltrate da cellule linfomonocitarie talora con aree di necrosi fibrinoide. Si possono verificare anche trombosi venose e meno frequentemente arteriose.
Storia

La malattia prende il nome da Hulusi Behçet (1889-1948), un dermatologo e scienziato turco che nel 1924 per primo riconobbe la condizione in uno dei suoi pazienti e che nel 1936 pubblicò i dati della sua scoperta sul Journal of Skin and Venereal Diseases. La denominazione «malattia di Behçet» è stata formalmente adottata al Congresso Internazionale di Dermatologia svoltosi a Ginevra nel settembre 1947.
Tuttavia, secondo alcuni autori è possibile che i sintomi di questa condizione fossero già stati descritti da Ippocrate di Coo nel V secolo a.C., nel suo Epidemion (libro 3, caso 7).Julius Hirschberg (1843-1925), nel suo testo di storia della oftalmologia risalente al 1899, riporta una citazione da Ippocrate e ammette di non essere riuscito a identificare la condizione che viene così descritta:
|
«Un'infiammazione acquosa degli occhi, cronica e dolorosa, associata alla presenza di escrescenze sulle palpebre, internamente ed esternamente, chiamate «fichi», che in molti casi distrugge la vista.» |
Altri utilizzano la locuzione «sindrome di Adamantiades» o «sindrome di Adamantiades-Behçet», riconoscendo il lavoro svolto dallo studioso greco Benediktos Adamantiades. Tuttavia, in base allo standard ICD-10 dell'Organizzazione Mondiale della Sanità, la terminologia corretta è «sindrome di Behçet».
Fra il 1991 e 1998, alcuni ricercatori medici arabi e sauditi diedero un contributo importante alla descrizione della «sindrome di neuro-Behçet», un coinvolgimento neurologico nella malattia di Behçet, considerata una delle manifestazioni più devastanti della condizione.
Epidemiologia
La sindrome di Behçet è rara negli Stati Uniti e in Europa ma è comune in Medio Oriente, il che suggerirebbe una possibile causa endemica presente nelle aree tropicali. Nella Turchia occidentale e meridionale la prevalenza è di 20-420 casi ogni 100 000 abitanti, nei territori degli altri paesi medio-orientali questi valori scendono a 2,1-19,5, mentre in Europa meridionale e del nord sono rispettivamente del 1,5-15,9 e 0,3-4,9. L'incidenza nel Nord Italia è in linea con quella dei paesi nordeuropei.
Si stima che circa 15-20 000 cittadini statunitensi abbiano ricevuto la diagnosi di questa malattia. In uno studio del 2009 è stata messa in evidenza una incidenza, negli Stati Uniti d'America, pari a circa 0,38 casi per 100 000 persone; nel Regno Unito si stimano 0,6 casi ogni 100 000 persone. A livello globale, i maschi sono colpiti più frequentemente delle femmine, ma vi sono aree geografiche in cui questa prevalenza non si verifica.
Tale condizione non è associata con i tumori e non vi sono certezze circa la correlazione con i vari tipi di tessuti, anche se a tal proposito le ricerche continuano. Inoltre essa non segue il modello usuale delle malattie autoimmuni. Tuttavia, uno studio ha rivelato una possibile correlazione con le allergie alimentari, in particolare per i prodotti lattiero-caseari.
I risultati di uno studio epidemiologico hanno mostrato come il 56% dei pazienti con malattia di Behçet abbia sviluppato coinvolgimento oculare a un'età media di 30 anni: ciò ha rappresentato la prima manifestazione della sindrome nell'8,6% dei pazienti. Un interessamento che si estende anche al nervo ottico è stato raramente segnalato. Tra i pazienti che hanno accusato problemi oculari, nel 17,9% dei casi è stata identificata una atrofia ottica, nel 9% una emorragia retinica, nel 13,3% un edema maculare mentre nel 5,8% una occlusione di un ramo di una vena retinica. L'atrofia ottica è stata individuata come la causa più importante di disabilità visiva ed è stata riscontrata nel 54% dei pazienti con malattia oculare di Behçet e deficit visivo permanente. Il decorso della malattia è più grave nei pazienti con un'età precoce di insorgenza e che presentano coinvolgimento gastrointestinale.
Fisiopatologia
Sebbene la causa della sindrome di Behçet non sia ancora ben definita, si ritiene che essa sia una malattia autoimmune, i cui fattori scatenanti possono essere di natura genetica, infettiva o immunitaria.
Genetica
La malattia di Behçet è certamente un disturbo sporadico, tuttavia è noto da tempo e probabilmente è causata da una mutazione del gene che contiene la proteina LACC1. La prevalenza della sindrome è maggiore nelle zone circostanti l'antica rotta commerciale della seta in Medio Oriente: per questo motivo alcuni studiosi si riferiscono a essa come alla «malattia della via della seta». Questo fatto potrebbe far pensare a un'eziologia genetica, malgrado la sindrome non colpisca esclusivamente individui provenienti da tale area geografica.
Molti studi sierologici hanno mostrato una correlazione fra la malattia e l'antigene HLA-B51. La presenza dell'HLA-B51 è più frequente nella popolazione del Medio Oriente: i soggetti con antigene HLA-B51 sono in maggioranza maschi e tendono a manifestare una più alta prevalenza di ulcere genitali, oculari e, in generale, di sintomi cutanei; al contrario il coinvolgimento gastrointestinale si verifica meno frequentemente. In ogni caso l'antigene HLA-B51 non ha mostrato di influenzare la gravità della sintomatologia.
Esposizione alle infezioni
L'esposizione a un agente infettivo può essere la causa scatenante di una risposta immunitaria cross-reattiva. Dal momento che la presenza ricorrente di ulcere orali costituisce un criterio fondamentale per la diagnosi della malattia, sono stati presi in esame i patogeni che vivono nella cavità orale, come il virus herpes simplex (HSV) e le specie batteriche Streptococcus, Staphylococcus ed Escherichia coli.
Molti studiosi si sono concentrati su un possibile coinvolgimento delle proteine umane da shock termico (heat shock proteins, o HSP, ovvero degli antigeni unici dotati di potente capacità immunostimolatrice), e in particolare su HSP-60 e HSP-65. È stato dimostrato che alcuni peptidi derivati da HSP-60 sono in grado di indurre una significativa proliferazione delle cellule T negli individui affetti dalla sindrome di Behçet. HSP-65 è stato ritrovato in alte concentrazioni in pazienti che presentano ulcere orali e lesioni cutanee attive. Questo antigene è stato individuato come un efficace stimolatore della produzione di anticorpi che presentano reattività crociata con alcune specie di streptococchi normalmente presenti nel cavo orale.
Sistema immunitario

La sindrome di Behçet presenta un coinvolgimento di molteplici organi: attraverso uno studio al microscopio si possono osservare delle infiltrazioni di cellule T e neutrofili nei tessuti colpiti.
Le analisi del sangue dei pazienti affetti dalla condizione mostrano una risposta predominante di linfociti T helper, sia CD4+ sia CD8+, con le corrispondenti elevazioni dei livelli di citochine (interleuchine e interferone-gamma).
Come detto, le lesioni caratteristiche della sindrome di Behçet vedono una forte attività e presenza delle cellule neutrofile. Ciò ha portato molti studi a indirizzarsi in tal senso; tuttavia, al 2013 non sono state ancora formulate teorie valide che caratterizzino il loro ruolo e i risultati si sono rivelati spesso contraddittori. Si ritiene, comunque, che il rilascio di citochine possa mettere i neutrofili in uno stato di «pre-eccitazione statico», grazie a un meccanismo non ancora ben determinato. Da questo stato i neutrofili possono essere poi innescati e portati in una condizione di iperattività a seguito di stimoli ambientali; ciò avviene, nei pazienti affetti dalla malattia, a una soglia inferiore rispetto agli individui che non ne soffrono.
Segni e sintomi

La malattia di Behçet è una patologia autoimmune, infiammatoria, sistemica, dalle cause ancora sconosciute, che coinvolge i vasi sanguigni (sia capillari, sia vene e arterie). I sintomi della malattia variano a seconda del distretto interessato, ma per formulare la diagnosi è necessaria una triade di sintomi osservati dal paziente o dal medico per più volte in un anno:
- afte orali;
- afte genitali;
- manifestazioni oculari come uveiti, neuriti ottiche, trombosi retinica, glaucoma.
La malattia di Behçet è una patologia cronica e con il tempo può colpire qualunque organo. Numerosi sintomi «minori», nel senso che hanno una frequenza inferiore, possono apparire in un momento o in un altro.
Sistema muscolo-scheletrico
Le artralgie infiammatorie, cioè i dolori alle articolazioni, sono una manifestazione molto comune, la terza in ordine di frequenza dopo l'interessamento mucocutaneo e le lesioni oculari: i disturbi articolari infatti possono colpire fino a oltre l'80% dei soggetti con sindrome di Behçet. Questi dolori sono spesso migranti e interessano principalmente le grandi articolazioni degli arti inferiori. Meno comunemente possono presentarsi anche manifestazioni di tipo artritico: secondo uno studio, l'oligoartrite ricorre nel 7,5% dei casi, la frequenza della monoartrite risulta leggermente inferiore (6,5%), mentre la poliartrite si manifesta nel 5% dei soggetti. Le articolazioni più colpite sono le ginocchia, le anche, i gomiti e i polsi. In un discreto numero di pazienti è possibile osservare un coinvolgimento dello scheletro assiale e il verificarsi di sacroileite (circa il 6-8%) e spondilite anchilosante (5% circa). Solitamente la patologia non produce nell'osso o nelle cartilagini danni permanenti: normalmente l'infiammazione resta circoscritta al collagene o al tessuto sotteso alle articolazioni. Forme insolite di interessamento del sistema muscolo-scheletrico includono l'artrite che si associa a malformazioni e/o a profonde alterazioni articolari, la pseudogotta, la rottura di cisti poplitea (con una clinica che imita un episodio di trombosi venosa profonda) e la miosite. Ovviamente l'artrite e le altre manifestazioni muscolo-scheletriche nei pazienti affetti da malattia di Behçet influenzano notevolmente il livello del dolore percepito e la qualità di vita.
Sistema gastrointestinale
Il coinvolgimento dell'apparato gastrointestinale è comune ed è caratterizzato da dolore addominale, flatulenza, diarrea e costipazione; spesso si ritrova anche muco o sangue nelle feci. Talvolta è possibile reperire lesioni a carico della mucosa ano-rettale: in una fase iniziale si manifestano ulcere localizzate e molto dolorose, che possono poi estendersi a interessare la mucosa dell'ampolla rettale e quindi del colon. Queste ulcere talvolta evolvono fino a formare delle vere e proprie fistole. Quando viene eseguita una colectomia e si procede ad analizzare il reperto anatomico, queste lesioni della mucosa appaiono, in genere, come ampie ulcerazioni spesso ad andamento longitudinale, associate a fessurazioni e vere e proprie afte. Le lesioni di solito si verificano all'interno di un contesto di mucosa normale o, al più, di infiammazione focale. La regola è riscontrare una associata vasculite linfocitaria che coinvolge le vene della sottomucosa.
Non è raro che, in assenza di altre localizzazioni o criteri di malattia, queste lesioni possano essere scambiate per manifestazioni proprie di una malattia infiammatoria intestinale cronica, come ad esempio la colite di Crohn. In ogni caso sono stati elaborati alcuni criteri endoscopici per differenziare i due diversi tipi di lesioni.
Rene, vie urinarie e annessi genitourinari
È stato rilevato che una lieve insufficienza renale si può presentare in una frazione variabile fra lo 0% e il 55% dei casi di sindrome di Behçet; il sesso maschile rappresenta un fattore di rischio per questa manifestazione clinica. Generalmente la condizione è asintomatica con rari casi di gravi coinvolgimenti renali; le cause più frequenti dell'insufficienza renale sono l'amiloidosi e la glomerulonefrite. Una diagnosi precoce può essere formulata grazie all'analisi delle urine, oltre alla misurazione della creatinina sierica e dei livelli di azoto ureico nel sangue.
Occasionalmente può presentarsi anche dolore e gonfiore a livello dei testicoli (epididimite) o dell'uretra (uretrite) o della prostata (prostatite).
Sistema cardiovascolare
L'infiammazione dei vasi può indebolire le pareti dei vasi stessi e provocare quindi un aneurisma. Nonostante si tratti di una complicanza rara, è comunque pericolosa. Queste manifestazioni vascolari sono dei buoni predittori di morbilità e mortalità dei pazienti con malattia di Behçet. Il coinvolgimento dei grossi vasi rappresenta una manifestazione comune della sindrome. Questi pazienti presentano comunemente flebiti ricorrenti, in genere a carico delle vene di maggiore diametro (ad esempio la vena cava superiore e inferiore, oppure le vene epatiche) così come delle vene cerebrali. Quando sono coinvolti i vasi arteriosi, generalmente la manifestazione di sofferenza è rappresentata dalla formazione di aneurismi o di pseudoaneurismi: con grande frequenza è possibile osservare l'interessamento dell'aorta ascendente (che spesso conduce a insufficienza aortica), aneurismi dell'aorta discendente, aneurismi dell'arteria polmonare, pseudoaneurismi dell'arco aortico, aneurismi dell'aorta toracica e addominale. Le complicanze vasali e arteriose rappresentano inoltre problemi di difficile soluzione chirurgica, in quanto ogni intervento invasivo sul sistema arterioso può condurre allo sviluppo di pseudoaneurismi, in considerazione della debolezza intrinseca delle pareti dei vasi. Le manifestazioni cardiologiche rappresentano eventi assolutamente non comuni in corso di malattia di Behçet ricorrendo in percentuale variabile, secondo diversi studi epidemiologici, fra il 7% e il 30% dei pazienti. Le principali manifestazioni sono rappresentate da pericarditi, endocarditi, miocarditi, fibrosi endomiocardica, vasculite delle arterie coronariche, trombosi intracardiaca, aneurisma del seno di Valsalva, disfunzioni delle valvole cardiache e disturbi del sistema di conduzione.
Sistema nervoso centrale

Nei casi avanzati della sindrome di Behçet può presentarsi un coinvolgimento a livello del sistema nervoso centrale, che rappresenta una delle manifestazioni più devastanti della malattia. Secondo alcuni studi, l'interessamento neurologico può interessare da poco più del 5% fino al 50% dei pazienti. Possono verificarsi lesioni parenchimatose (ad esempio le meningoencefaliti, che contraddistinguono il cosiddetto quadro «neuro-Behçet») e lesioni non parenchimatose (tipicamente le trombosi delle vene cerebrali e gli aneurismi delle arterie, che identificano il caso «vasculo-Behçet»). Nella maggior parte dei pazienti la malattia sembra comunque caratterizzarsi per l'interessamento infiammatorio-vascolare del sistema nervoso centrale (SNC), con interessamento parenchimale focale o multifocale, il quale tendenzialmente comporta l'insorgenza di una sindrome subacuta del tronco cerebrale e di emiparesi. Frequente è pure la comparsa di ipertensione endocranica (neuro-Behçet extrassiale).
Forti mal di testa sono sintomi sempre riscontrabili nella sindrome di Behçet, ma solo in pochi casi è sinonimo di interessamento cerebrale. Mal di testa particolarmente severi dovrebbero essere valutati con attenzione da un neurologo. Le trombosi nel cervello non sono comuni, ma comunque possibili, così come l'infiammazione delle meningi (la meningoencefalite). Questi processi infiammatori possono essere diagnosticati grazie a una risonanza magnetica che mostra immagini dell'organo e del suo sistema vascolare.
Cute, mucose e annessi cutanei
Le manifestazioni muco-cutanee della malattia sono estremamente comuni.
A livello cutaneo, la malattia può provocare lesioni simili a quelle proprie dell'eritema nodoso, follicolite e pseudo follicolite, lesioni simil acneiche in età adulta e altre che imitano quelle proprie del pioderma gangrenoso.
La vasculite a livello cutaneo può essere evidenziata dal fenomeno spontaneo della patergia (per punture di insetti o iniezioni). Il fenomeno può anche essere svelato e provocato dal medico tramite la somministrazione sottocutanea di una minima quantità di soluzione fisiologica: si tratta del test della patergia, un test non specifico di iper-reattività cutanea verso i minimi traumi, che a distanza di 24-48 ore dall'inoculazione intradermica di soluzione fisiologica comporta, nella sede di iniezione, la formazione di una papula arrossata, una vescicola o una pustola di diametro superiore ai 5 mm.
Manifestazioni oculari
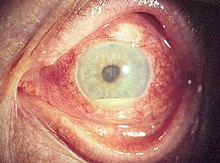
Le manifestazioni oculari nel corso della malattia sono estremamente comuni: le malattie infiammatorie dell'occhio possono svilupparsi precocemente nel corso della sindrome e portare alla perdita permanente della vista nel 20% dei casi. Il coinvolgimento oculare può verificarsi sotto forma di uveite posteriore, uveite anteriore o vasculite occlusiva retinica. I soggetti affetti da uveite anteriore si caratterizzano per la presenza di dolore e arrossamento oculare, ipopion sterile e riduzione dell'acuità visiva. Coloro che vengono colpiti da uveite posteriore presentano piuttosto una diminuzione dell'acuità visiva e corpi galleggianti nel campo visivo, ma il disturbo in genere è indolore. Una rara forma di coinvolgimento oculare in questa sindrome è data dalla vasculite retinica che si presenta con diminuzione indolore dell'acuità visiva e con la possibilità di presenza di corpi galleggianti o altri difetti del campo visivo.
Altre manifestazioni
Molto spesso sono presenti febbre alta intermittente all'esordio della malattia e febbricola serale nella fase cronica, così come una stanchezza marcatissima fin dal mattino (astenia). Studi clinici relazionano la riacutizzazione della malattia di Behcet, così come di tutte le malattie autoimmuni, con la presenza di fattori stressanti siano essi fisici o psicologici.
Le terapie necessarie (immunosoppressori, farmaci biologici) spesso rendono i pazienti più soggetti a stati influenzali o simil-influenzali.
Diagnosi
La sindrome di Behçet non è diagnosticabile né da sintomi patognomici (cioè manifestazioni associate univocamente alla malattia), né da esami di laboratorio. Per effettuare la diagnosi si procede dunque seguendo i criteri proposti nel 1990 dall'International Study Group for Behçet's disease.
Affinché si possa formulare una diagnosi di sindrome di Behçet, i pazienti devono presentare – in assenza di altri quadri clinici che giustifichino questi sintomi – un'ulcerazione orale ricorrente (aftosa o erpetiforme) osservata dal medico o dal paziente almeno tre volte in un periodo di 12 mesi con l'aggiunta di almeno due o più di questi criteri:
- ulcerazione genitale ricorrente;
- lesioni oculari:
- uveite anteriore;
- uveite posteriore (cellule del corpo vitreo osservate tramite lampada a fessura);
- vasculite retinica;
- lesioni cutanee:
- eritema nodoso;
- follicolite;
- lesioni papulo-pustolose o noduli acneiformi in pazienti in età post-adolescenziale che non assumano corticosteroidi: queste lesioni, nel caso in cui diano positività al test cutaneo alla patergia (fenomeno di iper-reattività cutanea aspecifica), indirizzeranno alla diagnosi entro 24-48 ore; tale test può risultare di qualche utilità nei soggetti che vivono nelle zone orientali estreme o centrali, ma nel mondo occidentale malattia di Behçet e patergia non rappresentano un'associazione consistente.
La positività dell'antigene B5-51 ha valore soltanto per confermare la diagnosi in caso si manifestino soltanto due dei tre sintomi necessari. La diagnosi precoce di queste forme è fondamentale per avviare la terapia idonea, che conduce spesso alla risoluzione della sintomatologia.
Diagnosi differenziale
Tra le varie diagnosi differenziali per la sindrome di Behçet figurano: amiloidosi, sindrome da anticorpi antifosfolipidi, malattie infiammatorie croniche intestinali, sindromi paraneoplastiche, poliarterite nodosa, lupus eritematoso sistemico.
Inoltre molte manifestazioni della malattia possono risultare simili a sindromi associate all'antigene HLA-B27 (come spondilite anchilosante, artrite psoriasica, artrite reattiva), agli stati di trombofilia, alle condizioni di malnutrizione, alla sarcoidosi e alle infezioni batteriche. Le ulcerazioni alla bocca possono essere associate anche con la malattia di Crohn, la dermatite bollosa, con l'HIV o con il deficit di vitamine. Ricorrenti uveiti possono presentarsi anche nella malattia di Crohn, nella sarcoidosi e nella sindrome di Vogt-Koyanagi-Harada. Le lesioni arteriose mimano gli effetti della poliarterite e della arterite di Takayasu.
Trattamento

Il trattamento ha lo scopo di alleviare i sintomi, riducendo l'infiammazione e controllando il sistema immunitario. Una terapia basata sulla somministrazione di alte dosi di corticosteroidi (1 mg/kg/die di prednisone orale) trova indicazione nelle manifestazioni più gravi della malattia. La terapia con inibitori del TNF-alfa, come l'infliximab, ha mostrato risultati promettenti nel trattamento dell'uveite associata con la malattia. Un altro farmaco anti TNF-alfa, l'etanercept, può essere utile nei pazienti con sintomi prevalentemente cutanei e delle mucose.
L'Interferone alfa-2a può rappresentare un efficace trattamento alternativo, in particolare per le ulcere genitali e orali e per le lesioni oculari. L'azatioprina, quando usata in combinazione con interferone alfa-2b presenta anch'essa risultati soddisfacenti, mentre la colchicina può essere utile per il trattamento di alcuni ulcere genitali, dell'eritema nodoso e dell'artrite.
Anche il talidomide è stato utilizzato per il suo effetto immuno-modificante. Il dapsone e il rebamipide, in studi su piccola scala, hanno mostrato risultati positivi per le lesioni mucocutanee.
Data la sua rarità, al 2013 non è stato ancora definito un trattamento ottimale per la neuropatia ottica acuta nella malattia di Behçet. L'identificazione e il trattamento precoce è tuttavia essenziale. È stata riportata una risposta alla ciclosporina, alla triamcinolone perioculare e al metilprednisolone IV seguiti da prednisone orale, tuttavia si è segnalata pure la possibilità di ricadute che possono portare alla perdita visiva irreversibile anche durante la cura. Gli immunosoppressori, come l'interferone alfa e gli inibitori del TNF-alfa possono migliorare ma non invertire completamente i sintomi oculari della Behçet, che potrebbe anche evolvere nel tempo malgrado il trattamento. Quando i sintomi sono limitati alla porzione anteriore dell'occhio, la prognosi è migliore; un coinvolgimento della porzione posteriore, e in particolare del nervo ottico, sono invece degli indicatori prognostici negativi. L'atrofia secondaria del nervo ottico è spesso irreversibile: una puntura lombare o il trattamento chirurgico possono essere richiesti per evitare l'atrofia ottica nei casi di ipertensione endocranica refrattaria al trattamento con immunomodulatori e steroidi.
La somministrazione di immunoglobuline endovena potrebbe essere un trattamento valido per i casi più gravi o complicati.
Prognosi
La prognosi per la sindrome di Behçet è correlata al sito di manifestazione e alla gravità del suo coinvolgimento. Tuttavia, la condizione aumenta significativamente la morbilità e la mortalità di chi ne soffre. In generale è possibile affermare che la malattia si manifesta in modo meno grave nel sesso femminile, e ciò sembra vero per ogni tipo di manifestazione clinica della sindrome. A titolo d'esempio, in uno studio del 2003 condotto su 387 pazienti (di cui 262 maschi e 125 femmine), nessuna donna colpita dalla malattia presentava aneurismi arteriosi.
Per evitare le manifestazioni della malattia, i pazienti possono necessitare dell'assunzione a vita di farmaci immunosoppressori, utilizzando comunque la dose più bassa possibile per evitare i loro gravi effetti collaterali.
Tra le complicanze più frequenti vi è la possibilità di incorrere in aneurismi, in eventi trombotici e vasculiti con conseguenti eventi ischemici distali, perdita della vista in seguito a uveite anteriore e posteriore, coinvolgimento neurologico. Secondo alcuni studi la causa principale di mortalità è la malattia dei grandi vasi, soprattutto l'emorragia dovuta alla rottura di aneurismi coronarici o dell'arteria polmonare, evento, quest'ultimo, registrato quasi esclusivamente tra i pazienti maschi. Altri studi mettono invece in evidenza l'elevata mortalità correlata al coinvolgimento neurologico.
Non risultano, al 2013, molte analisi che abbiano prodotto dati inerenti al follow-up del paziente. Uno studio effettuato in Giappone, su 2 031 pazienti a un anno dalla diagnosi, ha mostrato come il 31,7% avesse riportato un peggioramento della propria condizione clinica e lo 0,9% fosse deceduto.
In base a una ricerca del 2010 effettuata su 817 pazienti affetti dalla sindrome, 41 di essi (5%) erano deceduti dopo un follow-up di 7,7 anni; a 1 e 5 anni il tasso di mortalità era stato dell'1,2% e 3,3% rispettivamente, mentre l'età media dei decessi era stata di 34,6 anni con 11,5 anni di deviazione standard. Il 95,1% di loro era di sesso maschile. In quanto alle principali cause di morte, per il 43,9% erano legate a patologie dei vasi (aneurisma arterioso, in particolare delle arterie polmonari, e sindrome di Budd-Chiari), per il 14,6% le cause sono state il cancro e le malattie ematologiche maligne, infine per il 12,2% erano stati fatali il coinvolgimento del sistema nervoso centrale e la sepsi.
Bibliografia
- (EN) Stephen J. McPhee, Lawrence M. Tierney, Jr., Maxine A. Papadakis (a cura di), Current Medical Diagnosis and Treatment 2007 (Current Medical Diagnosis & Treatment), 46ª ed., McGraw-Hill Professional, 2006, ISBN 978-0-07-147247-0.
- (EN) Sungnack Lee, Dongsik Bang, Eun-So Lee, Behçet’s Disease: A Guide to Its Clinical Understanding, Springer, 2001, ISBN 978-3-540-66761-2.
- Ettore Bartoli, Medicina interna. Metodologia, semeiotica, fisiopatologia, clinica, terapia medica, RAHP sas, 2010, pp. 353-354, ISBN 978-88-904381-1-0.
- (EN) Yusuf Yazıcı, Hasan Yazıcı, Behçet’s Syndrome, Springer, 2010, ISBN 978-1-4419-5641-5.
- (EN) Shigeaki Ohno, Manfred Zierhut, Immunology of Behçet's Disease, CRC Press, 2013, ISBN 978-0-203-97106-2.
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su sindrome di Behçet
Wikimedia Commons contiene immagini o altri file su sindrome di Behçet
Collegamenti esterni
- SIMBA ONLUS – Associazione Italiana Sindrome e Malattia di Behcet, su behcet.it.
- Scheda Informativa sulla Malattia di Behçet – Centro di Coordinamento per le Malattie Rare – Regione Lombardia
| Controllo di autorità | NDL (EN, JA) 00560569 |
|---|