
| Sindrome di Guillain-Barré | |
|---|---|
| Specialità | neurologia |
| Classificazione e risorse esterne (EN) | |
| OMIM | 139393 |
| MeSH | D020275 |
| MedlinePlus | 000684 |
| eMedicine | 792008, 1169959, 315632 e 1180594 |
| Sinonimi | |
| Paralisi di Landry Sindrome di Guillain-Barré | |
| Eponimi | |
|
Georges Guillain Jean Barré | |
La sindrome di Guillain-Barré (pronuncia [ɡi'lɛ̃ baˈre]), chiamata a volte paralisi di Landry o sindrome di Guillain-Barré-Strohl, è una radicolo-polinevrite acuta (AIDP, acute inflammatory demyelinating polyradiculoneuropathy) che si manifesta con paralisi progressiva agli arti con andamento disto-prossimale (di solito prima le gambe e poi le braccia). Il suo esordio è alquanto rapido ed è causata dal sistema immunitario del paziente che attacca il proprio sistema nervoso periferico. I tipici sintomi sono modificazioni nella sensibilità o nella percezione del dolore e debolezza muscolare, a cominciare dai piedi e dalle mani. Ciò, spesso, si diffonde alle braccia e alla parte superiore del corpo, coinvolgendo entrambi i lati. I sintomi si sviluppano nell'arco di poche ore fino a qualche settimana. Durante la fase acuta, la malattia può essere pericolosa per la vita, con circa il 15% dei pazienti che sviluppa debolezza dei muscoli respiratori che richiedono il ricorso alla ventilazione meccanica. Alcuni possono sperimentare cambiamenti nella funzione del sistema nervoso autonomo, comportando anche pericolose anomalie nella frequenza cardiaca e nella pressione sanguigna.
La causa è sconosciuta. Il meccanismo sottostante comporta una malattia autoimmune in cui il sistema immunitario attacca erroneamente i nervi periferici e danneggia il loro isolamento mielinico. A volte questa disfunzione immunitaria è scatenata da un'infezione o, meno comunemente, da un intervento chirurgico. La diagnosi viene solitamente formulata sulla base di segni e sintomi, attraverso l'esclusione di cause alternative, e supportata da test come studi sulla conduzione nervosa (elettromiografia) e l'esame del liquido cerebrospinale. Esistono numerosi sottotipi della condizione, in base alle aree di debolezza, ai risultati degli studi sulla conduzione nervosa e alla presenza di determinati anticorpi. È classificato come una polineuropatia acuta.
In quelli con grave debolezza, un trattamento tempestivo con la somministrazione di immunoglobuline per via endovenosa o plasmaferesi, insieme a terapie di supporto, porterà nella maggioranza dei casi a una buona ripresa. Un pieno recupero potrebbe, comunque, richiedere settimane o anni. Circa un terzo accuserà una debolezza permanente. Il 20%, anche con trattamento, conserva a vita disturbi neuromuscolari legati alla GBS dopo la totale guarigione nervosa.
Globalmente, la morte si verifica in circa il 7,5% delle persone colpite. La sindrome di Guillain-Barré è rara con 1 o 2 casi per 100 000 individui all'anno, ma è la causa più comune di paralisi acuta non traumatica in tutto il mondo. Entrambi i sessi e tutte le zone del mondo presentano un'incidenza simile. La sindrome prende il nome dai neurologi francesi Georges Guillain e Jean Alexandre Barré, che la descrissero insieme al medico francese André Strohl nel 1916.
Storia

La prima descrizione della malattia fu compiuta nel 1859 dal medico francese Jean Landry, benché la sindrome sia stata probabilmente sempre presente nella storia umana. Secondo uno studio effettuato da Katherine Hall, ricercatrice della Dunedin School of Medicine dell'Università di Otago, pubblicato su “The Ancient History Bulletin”, a causare il decesso di Alessandro Magno potrebbe essere stata la sindrome di Guillain-Barré, anziché le febbri malariche o l'avvelenamento, tradizionalmente indicati, o l'abuso di alcolici o la pancreatite acuta come da molti ipotizzato in tempi moderni.
Nel 1916, Georges Guillain, Jean Alexandre Barré e André Strohl la diagnosticarono su due soldati e descrissero il metodo diagnostico - la dissociazione albumino-citologica - in un anomalo aumento della produzione di proteine nel liquido cefalorachidiano ma con una normale conta delle cellule. La sindrome è anche conosciuta come poliradicolonevrite acuta idiopatica, polinevrite idiopatica acuta, paralisi ascendente di Landry, poliradicoloneuropatia infiammatoria acuta demielinizzante.
Nel 1956, il neurologo canadese Miller Fisher descrisse la variante che porta il suo nome. mentre quattro anni prima, il neurologo britannico Edwin Bickerstaff, a Birmingham, osservò il tipo a encefalite del tronco cerebrale e insieme a Philip Cloake produsse ulteriori contributi nella descrizione della condizione in una pubblicazione del 1957. Già nel 1938, Guillain aveva comunque riportato alcune di queste caratteristiche prima della loro completa descrizione. Da allora sono stati descritti ulteriori sottotipi: la descrizione della neuropatia sensoriale assonale risale al 1990.
I criteri diagnostici sono stati sviluppati alla fine del 1970, dopo una serie di casi correlati a vaccinazione contro l'influenza suina, e sono stati perfezionati nel 1990. La definizione è stata rivista, nel 2009, dalla Brighton Collaboration (una istituzione per la sicurezza dei vaccini) ma ciò è stato fatto principalmente per la ricerca. La trasfusione di plasma è stata impiegata a partire dal 1978 e la sua utilità è stata confermata in studi più ampi effettuati nel 1985. La somministrazione endovenosa di immunoglobuline è stata praticata a partire dal 1988 e la sua non inferiorità rispetto alla plasmaferesi è stata dimostrata in studi realizzati nei primi anni 1990.
Secondo uno studio del 2003 la malattia che colpì nel 1921 il futuro Presidente degli Stati Uniti Franklin Delano Roosevelt, causandogli una disabilità permanente agli arti inferiori e un'aritmia cardiaca, non era la poliomielite ma la sindrome di Guillain-Barré.
Epidemiologia
La malattia ha un'incidenza annuale su scala mondiale di circa 0,6-4 casi su 100 000 individui, con l'Europa che si attesta a una media di 1,2-1,9 casi su 100 000, mentre la Cina presenta valori molto più bassi con circa 0,66 casi. Negli Stati Uniti d'America, la sindrome di Guillain-Barré è la più comune causa di paralisi flaccida. Gli uomini si ammalano 1,78 volte più delle donne. L'incidenza aumenta con l'età: vi è circa 1 caso ogni 100 000 persone con età inferiore ai 30 anni e circa 4 casi per 100 000 in quelli con più di 75 anni. Si è osservato un rischio maggiore tra il personale militare, probabilmente dovuto alla maggior probabilità di aver avuto antecedentemente un episodio di gastroenterite infettiva.
L'incidenza della sindrome di Guillain-Barré durante la gravidanza è di 1,7 casi per 100 000 e sono stati riportati anche casi neonatali congeniti.
Le varianti AMAN (acute motor axonal neuropathy) e AMSAN (acute motor-sensory axonal neuropathy) si verificano soprattutto nel nord della Cina, in Giappone e in Messico. In Cina, circa il 65% dei casi presenta una patologia assonale. Questo può essere correlato all'esposizione a diversi tipi di infezione, ma anche alle caratteristiche genetiche delle popolazioni. Si ritiene, infine, che la variante di Miller Fisher sia più comune nel Sud-est asiatico. Alcuni studi epidemiologici svolti in Giappone e in Bangladesh hanno evidenziato un'alta prevalenza di casi di sindrome di Guillain-Barré scatenati da un'infezione antecedente da Campylobacter jejuni (circa nel 69% dei casi) rispetto all'Europa e agli Stati Uniti.
Eziologia
Si ritiene che tutte le forme di sindrome di Guillain-Barré siano dovute a una risposta immunitaria ad antigeni estranei (ad esempio agenti infettivi) che colpisce erroneamente i tessuti nervosi, per via di un fenomeno chiamato mimetismo molecolare. Gli obiettivi di tale attacco immunitario si pensa possano essere i gangliosidi, presenti in grandi quantità nei tessuti nervosi periferici umani.
Due terzi delle persone con sindrome di Guillain-Barré hanno avuto un'infezione prima della comparsa della malattia, risalente a qualche giorno o qualche settimana prima. Più frequentemente essi sono episodi di gastroenterite o di una infezione delle vie respiratorie. In molti casi può essere confermata l'esatta natura dell'infezione e in circa il 30% di essi sono provocati dal batterio Campylobacter jejuni, che provoca la diarrea.
Un ulteriore 10% dei casi sono, invece, attribuibili al citomegalovirus (CMV, HHV-5). Nonostante ciò, solo pochissime persone con infezioni da Campylobacter o CMV sviluppano la sindrome di Guillain-Barré (rispettivamente 0,25-0,65 per 1 000 e 0,6-2,2 per 1 000 individui). Il ceppo di Campylobacter interessato può determinare il rischio di incorrere nella sindrome, diverse forme di batteri possiedono numerosi lipopolisaccaridi sulla loro superficie e alcuni possono indurre la condizione, mentre altri non sono in grado di farlo.
Correlazioni tra altre infezioni e sindrome di Guillain-Barré sono meno certe. Altri due herpesvirus (il virus di Epstein-Barr/HHV-4 e il virus varicella-zoster/HHV-3) e il batterio Mycoplasma pneumoniae sono stati associati con lo sviluppo della sindrome. Anche la febbre dengue virale tropicale e l'infezione da virus Zika sono state correlate a episodi di Guillain-Barré. Una precedente infezione da virus dell'epatite E è stata trovata per essere più comune nelle persone con la sindrome di Guillain-Barré.
Nonostante quanto detto, il 60% dei casi non ha una causa nota. In alcuni di essi si suppone che possa essere scatenata dal virus dell'influenza, oppure da una reazione immunitaria al virus. È stato rilevato un aumento dell'incidenza della sindrome di Guillain-Barré dopo la vaccinazione antinfluenzale effettuata durante la pandemia di influenza suina del 1976-1977. in cui si stimano 8,8 casi per milione di vaccinati che svilupparono complicazioni.
Da allora, un attento monitoraggio dei casi attribuibili alla vaccinazione ha dimostrato che l'influenza può provocare la sindrome di Guillain-Barré. Piccoli aumenti di incidenza sono stati osservati anche in successive campagne di vaccinazione, ma non nella stessa misura. Il vaccino contro la pandemia influenzale del 2009 (pandemia del virus H1N1/09) non ha causato un aumento significativo dei casi. Tuttavia, i benefici della vaccinazione contro l'influenza superano i piccoli rischi della sindrome di Guillain-Barré. Anche per chi ha già sperimentato la Guillain-Barré è considerato sicuro ricevere i vaccini antinfluenzali in futuro. Altri vaccini, come quelli contro la poliomielite, il tetano o il morbillo, non sono mai stati associati a un rischio di sviluppare la sindrome. Qualche caso di sindrome di Guillain-Barré è preceduto da neoplasie viscerali, linfomi, infezione da HIV o interventi chirurgici.
L’Agenzia europea del farmaco (EMA) ha inserito la sindrome di Guillain-Barrè come effetto "molto raro" del vaccino anti-COVID-19 di AstraZeneca.[3]
FDA, EMA e in Italia AIFA hanno inserito la sindrome di Guillain-Barrè tra gli effetti collaterali rari o molto rari del vaccino Janssen.
Il risultato finale di questo attacco autoimmune a carico dei nervi periferici, è un danno alla mielina, lo strato lipidico che isola il nervo. Ciò porta a un blocco di conduzione del nervo con conseguente paralisi muscolare che può essere accompagnata da disturbi sensoriali o del sistema autonomo.
Nei casi lievi, la funzionalità dell'assone (la lunga porzione assottigliata di un nervo responsabile della conduzione del segnale) rimane intatta e il recupero può essere rapido, grazie alla rimielinizzazione. Nei casi più gravi, si verifica danno assonale e il recupero dipende dalla rigenerazione di questo tessuto. Circa l'80% dei pazienti ha perdita di mielina, nel restante 20% vi è perdita patologica degli assoni.
La sindrome di Guillain-Barré, a differenza di patologie come la sclerosi multipla e la sclerosi laterale amiotrofica è un disturbo del nervo periferico e non causa, in generale, danni al cervello o al midollo spinale.
In aggiunta a quanto detto, la sindrome di Guillain-Barré può insorgere in pazienti oncologici in trattamento con nelarabina, giacché rappresenta uno degli effetti secondari di tale farmaco.
Fisiopatologia
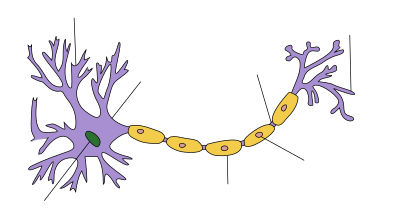
Nella sindrome di Guillain-Barré la disfunzione dei nervi è causata da un attacco immunitario alle cellule nervose del sistema nervoso periferico e alle loro strutture di supporto. Le cellule nervose hanno il loro corpo (soma) nel midollo spinale e una proiezione lunga (l'assone) che porta impulsi elettrici del nervo alla giunzione neuromuscolare dove l'impulso viene trasferito al muscolo. Gli assoni sono avvolti in una guaina di cellule di Schwann che contengono mielina. Tra le cellule di Schwann vi sono lacune (nodi di Ranvier) in cui è esposto l'assone. Alcune varianti cliniche della sindrome di Guillain-Barré presentano diversi tipi di attacco immunitario. La variante demielinizzante (AIDP) presenta un danneggiamento alla guaina mielinica perpetrato da parte dei globuli bianchi (linfociti T e macrofagi); questo processo è preceduto dall'attivazione di un gruppo di proteine del sangue noto come "sistema del complemento". Al contrario, la variante assonale è mediata dagli anticorpi IgG e dal sistema del complemento contro la membrana cellulare che copre l'assone senza un coinvolgimento diretto dei linfociti.
Sono stati riscontrati numerosi tipi di anticorpi in grado di colpire le cellule nervose. Nel sottotipo assonale, è stato dimostrato che questi anticorpi si legano ai gangliosidi, un gruppo di sostanze che si possono trovare nei nervi periferici. Un ganglioside è una molecola costituita da ceramide legata a un piccolo gruppo di zuccheri di tipo esoso e contenente diversi gruppi di acido N-acetilneuraminico. I principali quattro gangliosidi che vengono attaccati dagli anticorpi anti-gangliosidi sono GM1, GD1a, GT1a e GQ1b, con diversi anticorpi associati a particolari caratteristiche; ad esempio, gli anticorpi GQ1b sono stati correlati con la variante di Miller Fisher e le alcune forme simili come l'encefalite da Bickerstaff. Si ritiene che la produzione di questi anticorpi dopo un'infezione possa essere il risultato di un mimetismo molecolare in cui il sistema immunitario reagisce alle sostanze infettive microbiche ma gli anticorpi risultanti reagiscono anche con gli elementi presenti naturalmente nel corpo.
A seguito di un'infezione da Campylobacter, il corpo produce anticorpi della classe IgA; solo una piccola percentuale di persone produce anche anticorpi IgG destinati ad attaccare gli elementi della parete cellulare batterica (ad esempio lipopolisaccaride) che poi finiscono per colpire anche i gangliosidi delle cellule nervose dell'organismo. Non è attualmente noto come questo processo sfugga alla tolleranza centrale ai gangliosidi, il che è inteso a sopprimere la produzione di anticorpi contro l'organismo stesso. Non tutti gli anticorpi antigangliosidici causano la malattia e recentemente è stato suggerito che alcuni anticorpi si legano contemporaneamente a più di un tipo di epitopo (legame eterodimerico) e questo determina la risposta. Inoltre, lo sviluppo di anticorpi patogeni può dipendere dalla presenza di altri ceppi di batteri nell'intestino.
Segni e sintomi
I primi sintomi che appaiono nella sindrome di Guillain-Barré sono l'intorpidimento, la parestesia e il dolore, da soli o in combinazione. A questi seguono debolezza bilaterale delle gambe, che si manifesta come "gambe gommose" o gambe che tendono a deformarsi con o senza disestesie, e delle braccia; debolezza che tende a peggiorare con il tempo. Può essere necessaria da mezza giornata a oltre due settimane per raggiungere la massima gravità e poi quindi la situazione tende a stabilizzarsi. In un caso su cinque la debolezza continua a progredire per oltre quattro settimane. Anche i muscoli del collo possono essere colpiti e metà dei pazienti sperimenta un coinvolgimento dei nervi cranici, che innervano la testa e il viso; ciò può portare alla debolezza dei muscoli del volto, a difficoltà di deglutizione, scialorrea, difficoltà nella respirazione e, talvolta, a debolezza dei muscoli oculari.
Nell'8% dei pazienti la debolezza colpisce solo le gambe (paraplegia o paraparesi). Il coinvolgimento dei muscoli che controllano la vescica e l'ano è insolito. In totale, circa un terzo delle persone con la sindrome di Guillain-Barré continua a essere in grado di camminare. Una volta che la debolezza ha smesso di progredire, persiste a un livello stabile, "fase di plateau", prima che sopraggiunga un miglioramento. La fase di plateau può durare tra due giorni e sei mesi ma più comunemente persiste per una settimana. Più della metà dei pazienti accusa sintomi di dolore, che includono mal di schiena, parestesia dolorosa, dolori muscolari, mal di testa e mal di collo, relativi all'irritazione del rivestimento del cervello.
Molte persone con la sindrome di Guillain-Barré sperimentano i segni e i sintomi di una infezione, nelle 3-6 settimane prima della comparsa dei sintomi neurologici. Ciò può presentarsi come delle infezioni delle alte vie respiratorie (rinite, mal di gola) o diarrea.
Nei bambini, in particolare in quelli di età inferiore ai sei anni, la diagnosi può essere complicata e la condizione viene spesso inizialmente scambiata, talvolta fino a due settimane, per altre cause di dolori e difficoltà nella deambulazione, come infezioni virali o problemi all'apparato muscolo scheletrico.
All'esame neurologico, tratti caratteristici sono una potenza ridotta e riflessi tendinei ridotti o assenti, rispettivamente iporeflessia o areflessia. Tuttavia, una piccola percentuale presenta normali riflessi agli arti interessati, prima che si sviluppi l'areflessia, mentre alcuni possono avere riflessi esagerati. Nel sottotipo "variante Miller Fisher", la debolezza dei muscoli oculari (oftalmoplegia) è più marcata e può verificarsi con anomalie nella coordinazione (atassia). Il livello di coscienza normalmente non viene influenzato nella sindrome di Guillain-Barré, ma il sottotipo "encefalite del tronco cerebrale di Bickerstaff" può provocare sonnolenza o coma.
Forme lievi
Esistono anche delle forme lievi che rappresentano circa il 14% dei casi, senza paralisi totale e senza problemi respiratori, che andrebbero comunque monitorate e curate per evitare danni permanenti e indebolimento generale della muscolatura, con rischio di problemi muscoloscheletrici successivi, indebolimento dovuto alla denervazione e alla degenerazione walleriana; queste forme presentano, ad esempio, solo improvvisa difficoltà nella deambulazione (confondibile con la fatica da esercizio fisico intenso), che appare leggermente barcollante o claudicante ma è interamente conservata, con movimento dei piedi "a papera" (deambulazione anserina), oppure andatura steppante, a causa del coinvolgimento del nervo tibiale (pur senza arrivare al piede cadente) e del peroneo comune, prurito e debolezza muscolare agli arti superiori e conseguente difficoltà a svolgere alcune operazioni manuali, e leggere disfunzioni autonomiche. Medici di famiglia hanno riportato di numerosi pazienti che riferivano di prurito o lievi parestesie dopo infezioni alle vie aeree o intestinali; tali disturbi sono attribuibili a forme miti di GBS, spesso non diagnosticate.
In fase di recupero possibile presenza di fascicolazioni, mioclonie, crampi, tremore, affaticabilità ed esauribilità muscolare che possono divenire persistenti, come altre sequele neurologiche (cfr. Prognosi); nei pazienti con forme lievi la presenza di segni residuali permanenti è dovuta quasi sempre alla mancata diagnosi o al mancato trattamento, cosa che giustificherebbe l'uso delle immunoglobuline anche in questi casi. Spesso il paziente che ha avuto una GBS non grave e mostra affaticabilità persistente cronica dopo 6 mesi-1 anno, presenta però reperti elettroneurografici ed elettromiografici quasi normali alla media dei pazienti sani, in quanto la rigenerazione della guaina mielinica è comunque pressoché completa dopo questo periodo.
Insufficienza respiratoria
Un quarto di tutte le persone con la sindrome di Guillain-Barré sviluppa debolezza dei muscoli respiratori che portano a una insufficienza respiratoria e quindi all'incapacità di respirare adeguatamente per mantenere adeguati i livelli di ossigeno e/o anidride carbonica nel sangue. La maggior parte dei pazienti richiede un ricovero in ospedale e circa il 30% necessita di assistenza ventilatoria per il trattamento dell'insufficienza respiratoria tipo II. Questo scenario è pericoloso per la vita e rappresenta una complicanza se sussistono altri problemi medici come la polmonite, infezioni gravi, coaguli di sangue nei polmoni e emorragia gastrointestinale nel 60% di coloro che necessitano di ventilazione artificiale.
Disfunzione del sistema nervoso autonomo
Il sistema nervoso autonomo o involontario, che è coinvolto nel controllo delle funzioni fondamentali dell'organismo, come il mantenimento della frequenza cardiaca e della pressione sanguigna, viene influenzato nei due terzi dei casi di sindrome di Guillain-Barré, ma tale impatto è variabile. Nel 20% si riscontrano gravi fluttuazioni della pressione sanguigna e irregolarità nel battito cardiaco, a volte fino al punto che vi è la necessità di impianto di un pacemaker. Altri problemi correlati sono anomalie nella sudorazione (anidrosi, ipoidrosi o iperidrosi) e cambiamenti nella reattività delle pupille. Il coinvolgimento del sistema nervoso può colpire anche coloro che non hanno una grave debolezza muscolare.
Diagnosi

La diagnosi della sindrome di Guillain-Barré si effettua principalmente su base clinica e sul riscontro di alcuni segni, quali il rapido sviluppo della paralisi muscolare, l'assenza di riflessi, l'assenza di febbre e una causa probabile. Test diagnostici che possono essere effettuati per chiarire la condizione sono la rachicentesi (puntura lombare), con la quale si evidenzia un aumento delle proteine nel liquido spinale e delle cellule, ed esami elettromiografici e istologici dei nervi periferici.
L'esame del liquor, tramite puntura lombare, dimostra proteinorrachia non associata ad aumento delle cellule nel liquor (dissociazione albumino-citologica), ovvero un elevato livello di proteine con un basso numero di globuli bianchi; ciò può distinguere la sindrome da un certo numero di altre condizioni, come i linfomi e la poliomielite, dove sia le proteine sia il conteggio delle cellule sono elevati. Nella maggior parte dei casi di sindrome di Guillain-Barré, nel liquor si riscontra un quantitativo di proteine maggiore a 400 mg/L, con una conta delle normali cellule in un numero fisiologico. Nonostante ciò, l'esame del CSF nei primi giorni dall'esordio dei sintomi, è insignificante nel 50% delle persone e nell'80% dopo la prima settimana: pertanto, risultati normali non escludono la condizione. Non è raccomandabile una ripetizione della puntura lombare durante il corso della malattia, in quanto i livelli di proteina possono aumentare dopo l'inizio del trattamento.
All'esame istologico dei nervi periferici, ma soprattutto nelle fibre motorie e radici, si possono riscontrare infiltrati infiammatori costituiti da linfociti, monociti e macrofagi. si ritiene che la presenza di macrofagi possa essere responsabile della demielinizzazione.
La diagnosi dopo 5-6 giorni dall'esordio si dimostra con l'aumento dei tempi di conduzione, riduzione dei potenziali d'azione muscolare, le latenze distali sono prolungate così come l'onda F (interessamento porzioni prossimali del nervo) e onde H ritardate o assenti (perdita riflessi achillei). La valutazione diretta della conduzione nervosa degli impulsi elettrici può escludere altre cause di debolezza muscolare acuta, oltre a distinguere i diversi tipi di sindrome di Guillain-Barré. L'elettromiografia (EMG) e gli studi di conduzione nervosa possono essere eseguiti. Nelle prime due settimane, queste indagini potrebbero non mostrare alcuna anomalia. Gli studi di neurofisiologia, tuttavia, non sono necessari per raggiungere una diagnosi.
Anche il test per gli anticorpi anti gangliosidi viene spesso eseguito, ma il suo risultato non è determinante ai fini diagnostici. Le analisi del sangue vengono generalmente effettuate per escludere la possibilità di ulteriori motivi che spieghino la debolezza, ad esempio un basso livello di potassio nel sangue. Un anormalmente basso livello di sodio nel sangue si incontra spesso nella sindrome di Guillain-Barré. Ciò è stato attribuito alla inappropriata secrezione dell'ormone antidiuretico e alla relativa ritenzione di acqua.
L'imaging a risonanza magnetica risulta sensibile ma non specifico per la diagnosi, tuttavia, può essere un efficace strumento aggiuntivo, soprattutto per distinguere tra la sindrome di Guillain-Barré e altre condizioni che causano debolezza degli arti, come la compressione del midollo spinale.. Se la risonanza magnetica mostra un ispessimento delle radici dei nervi, questo può essere indicativo della sindrome di Guillain-Barrè. Tuttavia, nei bambini, ciò si riscontra nel 95% degli esami e quindi è necessario effettuare ulteriori esami.
Diagnosi differenziale
Tra le possibili diagnosi differenziali vi sono la miastenia gravis e polimiosite. Queste si distinguono per la flogosi, la distribuzione e la risposta alla terapia. Inoltre si può considerare il botulismo, la sindrome della cauda equina, l'intossicazione da metalli pesanti, malattia di Lyme, sclerosi multipla, neuropatia vasculitica, paralisi periodiche famigliari (es. paralisi periodica ipocaliemica) e miopatie metaboliche.
Varianti cliniche
Sono state riconosciute diverse varianti della sindrome. Esse le assomigliano per quanto riguarda l'evoluzione, i sintomi e la probabile patogenesi autoimmune, ma variano per quanto riguarda il recupero della salute.
- Poliradiculoneuropatia demielinizzante infiammatoria acuta (AIDP) è la forma che si riscontra più frequentemente negli Stati Uniti d'America. È generalmente preceduta da un'infezione batterica o virale. Quasi il 40% dei pazienti positivi per il batterio Campylobacter jejuni. Si riscontra una linfocitaria e macrofagica che causa la demielinizzazione dei nervi periferici. I sintomi generalmente si risolvono con la rimielinizzazione.
- Sindrome di Miller-Fisher (MFS): si riscontra in circa il 5% di tutti i casi di Guillain-Barré. Presenta i sintomi di atassia, areflessia e oftalmoplegia, quest'ultimo rappresenta una sua caratteristica cardinale. Il recupero avviene in genere in un periodo che varia da 1 a 3 mesi.
- Neuropatia assonale motoria acuta (AMAN): più frequente in età pediatrica. Caratterizzato da rapida progressione simmetrica e insufficienza respiratoria. Molti casi si sono verificati nelle zone rurali della Cina. La prognosi è generalmente buona con un recupero rapido, anche se in alcuni casi possono protrarsi delle disabilità anche per anni.
- Neuropatia sensoriale assonale e motoria acuta (AMSAN): è una forma più grave che colpisce anche i nervi sensoriali e le radici. I paziente sono solitamente adulti. Caratteristica è una marcata atrofia muscolare.
- Neuropatia acuta autonomica: è la variante più rara della sindrome. Coinvolge il sistema nervoso simpatico e parasimpatico. I pazienti accusano ipotensione posturale, anidrosi, diminuzione della salivazione e della lacrimazione e anomalie pupillari. Sintomi cardiovascolari sono comuni e le aritmie conseguenti possono essere causa di morte. Il recupero è graduale e talvolta non completo.
- Encefalite del tronco cerebrale di Bickerstaff: caratterizzata da una rapida insorgenza di areflessia e perdita sensoriale. La prognosi è solitamente buona. L'imaging a risonanza magnetica è fondamentale per la sua diagnosi.
In letteratura vi sono descritte altre varianti, ma esse rappresentano casi molto rari.
Trattamento
La terapia di supporto è la base di una buona gestione del paziente acuto affetto da sindrome di Guillain-Barré. Grande importanza è data al trattamento dell'insufficienza respiratoria dovuta alla paralisi del diaframma, il muscolo più importante per la respirazione. L'intubazione può rendersi necessaria quando vi è prova di danni a carico, quando la capacità vitale (VC) è minore di 20 mL/kg, la forza negativa inspiratoria (NIF) è maggiore (più vicino allo zero) rispetto -25 cmH2O, oltre il 30% di diminuzione della VC o della NIF entro 24 ore, nella rapida progressione di malattia o nel caso di instabilità autonomica.
Il successivo trattamento consiste nel tentare di ridurre l'attacco delle proprie difese immunitarie al sistema nervoso, mediante plasmaferesi, filtrando gli anticorpi dal flusso sanguigno o somministrando immunoglobuline per via endovenosa (IVIG), al fine di neutralizzare gli anticorpi nocivi e la malattia che causa l'infiammazione. Questi due trattamenti sono ugualmente efficaci e una combinazione dei due non è significativamente migliore della scelta di uno soltanto. La somministrazione di glucocorticoidi non si è dimostrata efficace per il trattamento della sindrome. Il trattamento viene generalmente cominciato subito dopo la diagnosi. La plasmaferesi accelera il recupero se viene intrapresa entro 4 settimane dalla comparsa dei sintomi. L'utilizzo di immunoglobuline ha un'efficacia equivalente alla plasmaferesi se cominciato entro 2 settimane dalla comparsa dei sintomi e presenta meno complicanze. Le immunoglobuline sono solitamente utilizzate come prima scelta, per la loro facilità di somministrazione e sicurezza. L'uso di immunoglobuline per via endovenosa non è esente da rischi, infatti possono, a volte, essere causa di epatiti e in rari casi, di insufficienza renale, se somministrate per più di cinque giorni.
Dopo la fase acuta, il trattamento consiste nella riabilitazione con l'aiuto di un team multidisciplinare che si concentra sul miglioramento delle attività della vita quotidiana (ADL). I terapisti occupazionali possono offrire attrezzature (ad esempio: sedie a rotelle e posate speciali) per aiutare il paziente a raggiungere l'indipendenza. I fisioterapisti possono aiutare a correggere il movimento funzionale, evitando compensazioni nocive che potrebbero avere un effetto negativo a lungo termine. Vi sono anche alcune prove a sostegno della fisioterapia per aiutare i pazienti con sindrome di Guillain-Barré a riguadagnare forza, resistenza e la qualità del passo, così come aiutarli a prevenire le contratture, le piaghe da decubito e i deficit cardiopolmonari. I logopedisti possono contribuire a recuperare la capacità di parlare e deglutire, specialmente se il paziente viene intubato o sottoposto a una tracheotomia.
Prognosi
Nella maggior parte dei casi, i pazienti affetti da sindrome di Guillain-Barré hanno un ottimo recupero. Esso solitamente comincia dopo la quarta settimana dal momento della comparsa del disturbo.
Circa l'80% dei pazienti ha un recupero completo in un tempo che va da pochi mesi a un anno,
Sequele neurologiche permanenti minori, come ad esempio l'areflessia, l'ipotonia muscolare o un lieve tremore possono verificarsi nel 10%-40% dei casi. Circa il 5-10% recuperano con gravi disabilità, in particolar modo con deficit motori prossimali e sensoriali, come: piede cadente bilaterale, atrofia muscolare, atassia e disestesia. Alcuni pazienti possono sviluppare la fibromialgia o la sindrome di Isaac, altri rigidezza e deformità osteoarticolare (es. piede cavo) e vertebrale, con successive infiammazioni radicolari o artrosiche.
Nonostante tutti i miglioramenti nella cura del trattamento e di supporto, il tasso di mortalità è ancora di circa il 2-3%, anche nel migliore dei reparti di terapia intensiva. Il tasso di mortalità su scala mondiale è leggermente superiore al 4%, principalmente per via della mancanza di disponibilità di apparecchiature di supporto vitale durante la fase critica, quando può essere necessario il ventilatore polmonare, nei peggiori casi. La principale causa di morte nei pazienti anziani con la sindrome di Guillain-Barré è il verificarsi di un'aritmia. Circa il 5-10% dei pazienti presenta una o più recidive tardive, nel qual caso vengono poi classificati come affetti da polineuropatia demielinizzante infiammatoria cronica.
Tra i fattori che possono peggiorare la prognosi vi sono: precedente infezione gastrointestinale, età avanzata, necessità di ventilazione meccanica, ricovero in terapia intensiva, scarsa terapia riabilitativa.
Stato della ricerca
La comprensione del meccanismo sottostante alla sindrome di Guillain-Barré si è evoluta negli ultimi anni. Lo sviluppo dei nuovi trattamenti è stato limitato dall'introduzione, tra gli anni 1980 e 1990, dell'immunoterapia. L'attuale ricerca ha lo scopo di dimostrare se alcune persone che hanno ricevuto immunoglobuline endovena possano trarre beneficio da un secondo ciclo di somministrazione, se i livelli di anticorpi nel sangue misurati dopo il trattamento hanno mostrato solo un piccolo aumento. Studi su farmaci immunosoppressivi come l'acido micofenolico, il fattore neurotrofico cerebrale e l'interferone beta (IFN-β) non hanno dimostrato benefici per sostenerne l'uso.
Su modelli animali di neurite autoimmune, alcuni agenti hanno mostrato risultati promettenti. Fra essi spiccano il glatiramer acetato, il quinpramine, il fasudil (inibitore dell'enzima Rho-chinasi) e la flecainide (antiaritmico di classe Ic).
Durante test di laboratorio, si sono dimostrati dei benefici con l'utilizzo di un anticorpo diretto contro l'anticorpo anti-ganglioside GD3. Dato il ruolo del sistema del complemento nella sindrome di Guillain-Barré, è stato suggerito che gli inibitori del complemento (come l'eculizumab) possono essere efficaci.
La sindrome di Guillain-Barré si presenta normalmente dopo un'infezione. Sono stati studiati vari casi di Guillain-Barré associati all'infezione da SARS-CoV-2 ma da primi studi su pochi casi non è stato possibile né determinare una causalità specifica né se ci sono manifestazioni tipiche della sindrome di Guillain-Barré associata a COVID-19 .
Bibliografia
- (EN) Gareth John Parry, Joel S. Steinberg, Guillain-Barre Syndrome: From Diagnosis to Recovery, a cura di American Academy of Neurology, Demos Medical Publishing, 2007, ISBN 978-1-932603-56-9.
- (EN) Silvia Iannello, Guillain-Barre Syndrome: Pathological, Clinical, and Therapeutical Aspects, Nova Publishers, 2004, ISBN 978-1-59454-170-4.
Voci correlate
Altri progetti
-
 Wikimedia Commons contiene immagini o altri file su sindrome di Guillain-Barré
Wikimedia Commons contiene immagini o altri file su sindrome di Guillain-Barré
Collegamenti esterni
- (EN) Sindrome di Guillain-Barré, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.
|
Classificazione e risorse esterne (EN) |
ICD-10-CM: G61.0; OMIM: 139393; MeSH: D020275; DiseasesDB: 5465;
MedlinePlus: 000684;eMedicine: 792008, 1169959, 315632 e 1180594; |