
| Malattia di Huntington | |
|---|---|
|
| |
| Malattia rara | |
| Cod. esenz. SSN | RF0080 |
| Specialità | neurologia |
| Eziologia | trinucleotide repeat expansion |
| Classificazione e risorse esterne (EN) | |
| ICD-9-CM | 333.4333.4,294.1294.1 |
| ICD-10 | G10 e F02.2 |
| OMIM | 143100 |
| MeSH | D006816 |
| MedlinePlus | 000770 |
| eMedicine | 1150165, 289706 e 792600 |
| GeneReviews | Panoramica e Panoramica |
| Sinonimi | |
|
Còrea di Huntington Chorea maior Morbo di Huntington | |
| Eponimi | |
| George Huntington | |
La malattia di Huntington, o còrea di Huntington (chorea major), è una malattia genetica neurodegenerativa che colpisce la coordinazione muscolare e porta a un declino cognitivo e a problemi psichiatrici. Esordisce tipicamente durante la mezza età; è la più frequente malattia a causa genetica nei quadri clinici neurologici con movimenti involontari anomali (che prendono il nome di corea).
È molto più comune nelle persone di discendenza europea occidentale che in quelle di origine asiatica o africana. La malattia è causata da una mutazione autosomica dominante in una delle due copie (alleli) di un gene codificante una proteina chiamata huntingtina, il che significa che ogni figlio di una persona affetta ha la probabilità del 50% di ereditare la condizione. La base genetica della malattia è stata scoperta nel 1993 grazie a una ricerca internazionale guidata dalla Hereditary Disease Foundation. I sintomi fisici della malattia possono incominciare a qualsiasi età, ma più frequentemente tra i 35 e i 44 anni.
I sintomi della malattia possono variare tra gli individui e anche tra i membri colpiti della stessa famiglia, ma di solito la loro progressione può essere predetta. I primi sintomi sono spesso sottili problemi di umore o cognitivi a cui segue una generale mancanza di coordinazione e un'andatura instabile. Con l'avanzare della malattia i movimenti non coordinati del corpo diventano sempre più evidenti e sono accompagnati da un calo delle capacità mentali e problemi comportamentali e psichiatrici. Le complicanze, come la polmonite, le malattie cardiache e i danni fisici da cadute, riducono l'aspettativa di vita a circa 20 anni a partire dall'esordio dei sintomi. Non esiste una cura per la condizione e un'assistenza a tempo pieno diventa necessaria nelle fasi più avanzate della malattia. I trattamenti sono solo farmacologici e non possono alleviare molti dei suoi numerosi sintomi.
Storia

Sebbene la corea di Huntington fosse stata riconosciuta come un disturbo fin da almeno il Medioevo, la causa è rimasta sconosciuta fino agli ultimi decenni del XX secolo. La condizione, nel corso degli anni, è stata associata a diversi appellativi, in origine chiamata semplicemente "corea" per i tipici movimenti a scatti danzanti, essa è stata poi anche chiamata "corea ereditaria" e "corea cronica progressiva".
La prima menzione certa della malattia, appare in una lettera di Charles Oscar Waters, pubblicata nella prima edizione del trattato Practice of Medicine di Robley Dunglison, nel 1842. Waters parlò di "una forma di corea, volgarmente chiamato magrums", aggiungendo descrizioni accurate dei sintomi, della loro progressione e la caratteristica ereditarietà della malattia.
Nel 1846 Charles Gorman osservò che in regioni ben localizzate vi era una più elevata prevalenza. Indipendentemente da Gorman e Waters, entrambi studenti di Dunglison al Jefferson Medical College di Filadelfia, anche Johan Christian Lund nel 1860 descrisse la condizione. Egli in particolare osservò che presso Setesdalen, una valle isolata di montagna in Norvegia, vi era un'elevata prevalenza di demenza associata a una condizione di disturbi del movimento che si riscontrava nelle famiglie.
La prima descrizione completa della malattia fu per opera di George Huntington e avvenne nel 1872. Esaminando la storia medica combinata di diverse generazioni di una famiglia che presentavano sintomi simili, egli si rese conto che queste condizioni dovevano essere correlate.
|
«Of its hereditary nature. When either or both the parents have shown manifestations of the disease ..., one or more of the offspring almost invariably suffer from the disease ... But if by any chance these children go through life without it, the thread is broken and the grandchildren and great-grandchildren of the original shakers may rest assured that they are free from the disease.» |
|
«Della sua natura ereditaria. Quando uno o entrambi i genitori hanno manifestato la malattia..., uno o più figli ne soffrono quasi invariabilmente... Ma se per caso questi figli non ne soffrono, il filo si spezza e i nipoti e i pronipoti di chi manifestò i sintomi possono essere certi di essere esenti dalla malattia.» |
Anche in Europa la malattia suscitò grande interesse tra gli scienziati, tra cui Louis Théophile Joseph Landouzy, Désiré-Magloire Bourneville, Camillo Golgi e Jules Déjerine, tanto che alla fine del XIX secolo gran parte della ricerca sulla malattia era europea. Verso la fine del XIX secolo, la malattia era ormai conosciuta in tutto il mondo come una condizione a sé stante.
Durante la riscoperta della teoria mendeliana, avvenuta a cavallo del XX secolo, la malattia di Huntington è stata utilizzata come esempio di trasmissione autosomica dominante. Il biologo inglese William Bateson utilizzò l'albero genealogico delle famiglie colpite per determinare l'eziologia genetica della condizione.
La ricerca per la causa della malattia ha avuto un forte progresso nel 1968, quando Milton Wexler, psicoanalista di Los Angeles, fondò la Hereditary Disease Foundation, dopo che alla moglie Leonore Sabin era stata diagnosticata la malattia di Huntington. Grazie a questa fondazione è stato avviato il progetto USA-Venezuela Huntington's Disease Collaborative Research Project nel 1979, che ha registrato un importante passo avanti nel 1983 con la scoperta della posizione approssimativa di un gene causale. Questo risultato è stato raggiunto grazie a uno studio che si è concentrato sulle popolazioni di due isolati villaggi venezuelani, Barranquitas e Lagunetas, dove c'era una prevalenza della malattia insolitamente alta.
Lo sviluppo di metodi per la marcatura del DNA ha reso possibile l'avvio del Progetto genoma umano. Nel 1993, il gruppo di ricerca ha isolato il gene causale preciso 4p16.3, rendendolo il primo locus riconosciuto utilizzando questo metodo di una malattia autosomica. Nello stesso tempo, vennero fatte scoperte fondamentali riguardanti i meccanismi della malattia, compresi gli accertamenti eseguiti dal gruppo di ricerca di Anita Harding sugli effetti della lunghezza del gene. La condizione originariamente chiamata corea di Huntington è conosciuta come malattia di Huntington, poiché non tutti i pazienti sviluppano la corea e per via dell'importanza dei problemi cognitivi e comportamentali.
Epidemiologia
L'insorgenza tardiva della malattia fa sì che essa solitamente non pregiudichi la riproduzione. La prevalenza in tutto il mondo della malattia di Huntington è di 5-10 casi ogni 100 000 persone, ma essa presenta una forte variazione geografica per via dell'etnia e delle migrazioni. La prevalenza è simile per uomini e donne. Il tasso di incidenza è più alto nelle popolazioni di discendenza europea occidentale, con una media intorno a 3-7 casi per 100 000 di persone, ed è più bassa nel resto del mondo, ad esempio 1 su 1 000 000 nelle persone di origine asiatica o africana. Inoltre in alcune aree ben localizzate si riscontra una prevalenza molto più elevata della media regionale.
Uno dei più alti valori di prevalenza, si registra nelle isolate popolazioni della regione del lago di Maracaibo in Venezuela, dove la malattia di Huntington colpisce fino a 7 000 individui su 1 000 000. Altre aree ad alta prevalenza sono state trovate in Tasmania e in specifiche regioni di Scozia, Galles e Svezia. L'aumento della prevalenza in alcuni casi si verifica a causa di un locale effetto del fondatore, dovuto a una storia di migrazione dei vettori in una zona di isolamento geografico. Analizzando studi genealogici alcuni di questi vettori sono stati fatti risalire a centinaia di anni fa.Aplotipi genetici possono anche fornire prove per le variazioni geografiche di prevalenza.
Fino alla scoperta del test genetico, le statistiche potevano includere soltanto la diagnosi clinica basata sui sintomi fisici e su una storia familiare di malattia di Huntington, escludendo coloro che erano morti per altre cause prima della diagnosi. Con la sempre maggior disponibilità del test genetico è probabile che i dati sull'incidenza e sulla prevalenza possano aumentare.
Eziologia
La malattia di Huntington è una delle malattie da espansione di triplette: esse sono causate dall'allungamento, in misura superiore al normale, di una sezione ripetuta di un gene. Il gene HTT è situato sul braccio corto del cromosoma 4 sul locus 4p16.3.
L'HTT contiene una sequenza di tre basi di DNA, citosina-adenina-guanina (CAG), ripetuta più volte e nota come espansione di triplette. CAG è il codice genetico per la codifica della glutammina, per cui una serie di queste triplette, porta alla produzione di una catena di glutammina nota come tratto poliglutamminico (o tratto polyQ), mentre la parte ripetuta del gene prende il nome di regione PolyQ.
Generalmente le persone sane presentano meno di 36 ripetizioni CAG nella regione polyQ; una sequenza di 36 o più triplette comporta la produzione di una proteina che ha caratteristiche diverse.
Ereditarietà

La malattia di Huntington è una malattia a ereditarietà autosomica dominante, il che significa che un individuo affetto, generalmente, eredita una copia del gene con un'espansione della ripetizione della tripletta nucleotidica (l'allele mutante) da un genitore affetto. La penetranza della mutazione è molto alta, coloro che possiedono una copia mutata del gene avranno la malattia. In questo tipo di modello di ereditarietà, ogni discendente di un individuo affetto avrà un rischio del 50% di ereditare l'allele mutante e quindi di essere colpito dalla malattia. Questa probabilità è indipendente dal sesso.
Patogenesi
La proteina huntingtina
Tutti gli esseri umani hanno due copie del gene che codifica la proteina huntingtina (HTT); il gene è chiamato anche IT15 (Interesting Transcript 15). Parte di questo gene è costituita da una sezione che varia in lunghezza tra individui e tra una generazione e l'altra. Quando la lunghezza di questa sezione ripetuta raggiunge una certa soglia, produce una forma alterata della proteina, chiamata proteina huntingtina mutata (mHtt, Htt mutante). Le funzioni differenti di questa proteina sono la causa delle alterazioni patologiche che a loro volta causano i sintomi della malattia. La proteina huntingtina (HTT) interagisce con oltre 100 altre proteine e sembra avere molteplici funzioni biologiche.
La mutazione della malattia di Huntington è geneticamente dominante a penetranza quasi completa: la mutazione di uno dei geni HTT di una persona causa la malattia. L'ereditarietà non è influenzata dal sesso, ma la lunghezza della sezione ripetuta del gene (e quindi la gravità) può essere influenzata dal sesso del genitore affetto: l'amplificazione della mutazione si verifica durante la spermatogenesi, ed è all'origine del fenomeno della anticipazione della malattia, se trasmessa da parte paterna.
| Numero di ripetizioni | Classificazione | Gravità della malattia | Rischio per la progenie |
|---|---|---|---|
| <26 | Normale | Non si svilupperà | Nessuno |
| 27–35 | Intermedio | Non si svilupperà | Elevato ma <<50% |
| 36–39 | Penetranza ridotta | Potrà o non potrà svilupparsi | 50% |
| 40+ | Penetranza completa | Si svilupperà | 50% |
La forma alterata mHtt aumenta la morte di alcuni tipi di neuroni. Diverse regioni del cervello presentano una differente quantità e dipendenza da questi neuroni e perciò il loro interessamento varia di conseguenza. Un numero di ripetizioni che varia da 36 a 39 porta a una malattia in forma di penetranza ridotta, con un'insorgenza molto più tarda di età e con una progressione più lenta. In alcuni casi l'esordio può essere così in ritardo che i sintomi non vengono mai notati. Con sequenze ripetute molte volte, la malattia ha una penetranza completa e può esordire anche al di sotto dell'età di 20 anni, venendo così indicata come malattia di Huntington giovanile o variante Westphal. Ciò rappresenta circa il 7% delle condizioni.
Ripetizioni della tripletta CAG superiori alle 28 risultano instabili nella fase di replicazione e questa instabilità tende ad aumentare all'aumentare del numero di ripetizioni presenti. Ciò porta spesso a nuove espansioni con il passaggio generazionale (mutazioni dinamiche) invece di riprodurre una copia esatta della ripetizione trinucleotide. Ciò comporta una modifica nel numero di ripetizioni presenti nelle generazioni successive, così che un genitore non affetto ma che presenta un numero "intermedio" di ripetizioni (28-35) o una "penetranza incompleta" (36-40), può trasmettere una copia del gene con un numero aumentato di ripetizioni arrivando ad avere una condizione di penetrazione totale della malattia.
Tali aumenti nel numero di ripetizioni (che comportano un abbassamento nell'età di insorgenza e un aumento della gravità della malattia) in successive generazioni è un fenomeno noto come anticipazione genetica. L'instabilità appare maggiore nella spermatogenesi rispetto alla oogenesi, infatti gli alleli ereditati dalla madre presentano una lunghezza di ripetizione simile, mentre quelli ereditati dal padre hanno una maggiore probabilità di presentare una lunghezza aumentata. È raro che la malattia di Huntington sia causata da una nuova mutazione in cui nessuno dei genitori possiede più di 36 ripetizioni CAG.
Il comportamento di questa proteina mutata non è completamente noto, ma si sa che è tossico per alcuni tipi di cellule, in particolare quelle del cervello. Il danno precoce è più evidente nel corpo striato, ma come la malattia progredisce, altre aree del cervello vengono colpite anche più vistosamente. I primi sintomi sono attribuibili alle funzioni del corpo striato e le sue connessioni corticali, val a dire il controllo sul movimento, l'umore e le funzioni cognitive superiori.
Nei rari casi in cui entrambi i genitori hanno un gene della huntingtina espanso, il rischio aumenta al 75%, e quando uno dei due genitori possiede due copie espanse, il rischio è al 100% (tutta la prole sarà interessata). Gli individui con entrambi i geni coinvolti sono rari. Per qualche tempo, la malattia di Huntington, è stata ritenuta l'unica condizione per la quale il possesso di un secondo gene mutato non influenzava né i sintomi né la progressione, ma da allora è stato scoperto che ciò può influenzare sia il fenotipo sia la velocità di progressione della malattia.
Funzione del gene della huntingtina
Il gene della huntingtina viene espresso in tutte le cellule di mammifero. Le concentrazioni più elevate si trovano nel cervello e nei testicoli, mentre vi sono moderate quantità nel fegato, nel cuore e nei polmoni. La funzione della proteina HTT nell'uomo non è chiara. Essa interagisce con le proteine che sono coinvolte nella trascrizione, nella segnalazione cellulare e nel trasporto intracellulare. Negli animali geneticamente modificati al fine di sviluppare la malattia di Huntington, sono state trovate diverse funzioni della HTT. In questi animali la proteina HTT si è dimostrata fondamentale per lo sviluppo embrionale, tanto che la sua assenza è legata alla morte dell'embrione. Essa agisce anche come agente anti-apoptotico per prevenire la morte cellulare programmata e controlla la produzione del fattore neurotrofico cerebrale, una proteina che protegge i neuroni e regola la loro nascita durante la neurogenesi.
L'HTT facilita anche il trasporto vescicolare, la trasmissione sinaptica neuronale e controlla la trascrizione genetica. Se l'espressione del gene HTT aumenta, viene di conseguenza aumentata la produzione della proteina HTT e la sopravvivenza delle cellule cerebrali risulta migliorata mentre gli effetti di mHTT sono ridotte. Quando invece l'espressioni del gene HTT sono ridotte, il risultato che si ottiene è tipico della presenza di mHTT. Nell'uomo, la distruzione del gene normale non causa la malattia. Si ritiene che la malattia non sia causata dalla produzione inadeguata di HTT, ma da un guadagno della funzionalità della mHTT.
La proteina CREB-binding (CBP)
La proteina CREB-binding (CBP), un fattore di trascrizione, risulta essenziale per la funzionalità cellulare, poiché agisce come co-attivatore per un numero significativo di promotori. Inoltre, gli amminoacidi che formano la CBP comprendono una serie di 18 glutammine. Così, le glutammine presenti sulla CBP interagiscono direttamente con l'aumento del numero di glutammine sulla catena HTT, questo comporta che la CBP viene spostata dalla sua tipica posizione vicino al nucleo. Nei cervelli di coloro che hanno avuto la malattia di Huntington, sottoposti ad autopsia, sono stati inoltre trovati avere una ridottissima quantità di CBP. Inoltre, quando vi è una sovraespressione di CBP, la morte cellulare indotta dalle poliglutamine risulta diminuita, dimostrando ulteriormente che la CBP gioca un ruolo importante nella malattia di Huntington e nel funzionamento in generale dei neuroni.
Complicanze
La maggior parte delle complicanze pericolose per la vita deriva dal coordinamento muscolare e, in misura minore, dai cambiamenti comportamentali indotti dal progressivo declino della funzionalità cognitiva. Il più grande rischio è di sviluppare la polmonite; metà degli affetti da malattia di Huntington muore per questa causa. Con il deterioramento della capacità di sincronizzare i movimenti, si presentano problemi di compensazione polmonare, un aumento del rischio di aspirazione di cibo o bevande e un incremento delle probabilità di contrarre la polmonite. Il secondo rischio più grande sono le malattie cardiache, che sono responsabili di quasi un quarto dei decessi di persone con la malattia. Il suicidio è la terza causa principale di decesso, con il 7,3% degli affetti da malattia di Huntington che si toglie la vita, mentre almeno il 27% tenta di farlo. Non è chiaro fino a che punto i pensieri suicidi siano influenzati da sintomi psichiatrici oppure dal desiderio del malati di evitare le fasi successive della malattia. Altri rischi associati comprendono soffocamento, lesioni da cadute e malnutrizione.
Anatomia patologica
Alterazioni cellulari
Vi sono molteplici cambiamenti cellulari attraverso cui la funzione tossica dell'mHTT (huntingtina mutata) può manifestarsi e quindi portare alla malattia di Huntington. Durante la modificazione post traduzionale dell'mHTT, il clivaggio della proteina può lasciare dietro piccoli frammenti costituiti da parte dell'espansione della poliglutamina. La natura polare della glutammina provoca interazioni con altre proteine quando vi è una sovrabbondanza di proteine HTT. Così, le molecola di mHTT formeranno legami idrogeno tra loro, formando una proteina aggregata anziché ripiegare a proteine funzionali. Nel tempo, gli aggregati si accumulano, incominciando a interferire con la funzione del neurone confluendo in un processo chiamato aggregazione proteica, una forma di corpi inclusi.
L'eccesso di proteine aggregate intorno agli assoni e ai dendriti porta all'interruzione della trasmissione dei neurotrasmettitori poiché le vescicole (piene di neurotrasmettitori) non possono più muoversi attraverso il citoscheletro. Questo processo porta, nel tempo, a una sempre minor capacità di rilascio dei neurotrasmettitori. I corpi inclusi sono stati trovati sia nel nucleo cellulare sia nel citoplasma. La presenza dei corpi di inclusione nei neuroni, sono uno dei primi cambiamenti patologici e alcuni esperimenti hanno dimostrato che possono essere tossici, tuttavia altri esperimenti hanno invece provato che essi possono costituire una parte del meccanismo di difesa aiutando nella protezione della cellula.
Sono stati identificati diversi modi per cui l'mHTT può causare la morte cellulare. Questi includono: effetti sulle proteine chaperone, che aiutano le proteine a ripiegare e a rimuovere quelli mal ripiegate; interazioni con le caspasi, che rivestono un ruolo nel processo di rimozione delle cellule; l'effetto tossico della glutammina sulle cellule nervose; la menomazione della capacità di produzione di energia all'interno delle cellule; effetti sull'espressione di geni. Gli effetti citotossici della mHTT sono fortemente amplificati dalle interazioni con una proteina chiamata Rhes, che si esprime principalmente nel corpo striato.
Alterazioni macroscopiche

La malattia di Huntington colpisce tutto il cervello, ma alcune aree appaiono più vulnerabili di altre. Gli effetti precoci più importanti si verificano soprattutto in una zona dei nuclei della base, chiamata corpo striato che è composta dal nucleo caudato e dal putamen. Altre zone colpite sono la substantia nigra, le aree 3, 5 e 6 della corteccia cerebrale, l'ippocampo, le cellule di Purkinje nel cervelletto, i nuclei tuberali laterali dell'ipotalamo e parti del talamo. Queste zone sono interessate per via della loro struttura e della tipologia di neuroni che esse contengono. Quando esse perdono cellule, appaiono ridotte di dimensioni. I neuroni spinosi dello striato sono i più vulnerabili, in particolare quelli che presentano proiezioni verso il globo pallido esterno, mentre gli interneuroni e le cellule spinose con proiezioni nel globo pallido interno sono meno colpiti. La malattia di Huntington provoca anche un aumento anomalo negli astrociti e l'attivazione delle microglia, le cellule immunitarie del cervello.
I nuclei basali, la parte del cervello più ampiamente colpita nelle fasi iniziali della malattia, giocano un ruolo chiave nel movimento e nel controllo del comportamento. Le loro funzioni non sono pienamente comprese, ma le teorie correnti suggeriscono che essi facciano parte del sistema delle funzioni esecutive e del circuito motorio. I nuclei della base normalmente inibiscono un gran numero di circuiti che generano movimenti specifici. Per intraprendere un movimento particolare, la corteccia cerebrale invia un segnale ai nuclei basali che porta a inibire uno specifico circuito e quindi a effettuare il movimento. Danni ai nuclei basali possono causare il rilascio o il ripristino delle inibizioni in un modo irregolare e incontrollato, ciò si traduce in movimenti incominciati involontariamente o interrotti prima del loro completamento voluto. I danni accumulati in questa zona provocano così i caratteristici movimenti correlati alla malattia di Huntington.
Clinica

Una visita medica, talvolta associata a un esame psichiatrico, può determinare se è incominciata l'insorgenza della malattia. Il verificarsi di eccessivi movimenti accidentali, di qualsiasi parte del corpo, sono spesso la ragione per cui viene intrapresa la consultazione medica. Se questi movimenti appaiono bruschi e con una distribuzione casuale, essi suggeriscono una diagnosi di malattia di Huntington. Sintomi cognitivi o psichiatrici sono raramente presenti alla prima diagnosi, mentre sono solitamente riconosciuti a posteriori o quando si sviluppano ulteriormente. Il punto di progressione della malattia può essere misurato usando la scala di valutazione "Unified Huntington" che fornisce un sistema generalizzato di valutazione basata sulla funzionalità motoria, comportamentale, cognitiva e funzionale.
Segni e sintomi
I sintomi della malattia di Huntington generalmente diventano evidenti tra i 40 e i 50 anni, ma possono esordire a qualsiasi età, dall'infanzia alla vecchiaia. Nelle prime fasi si riscontrano lievi cambiamenti nella personalità, nelle facoltà cognitive e nell'abilità fisica. I sintomi fisici sono solitamente i primi a farsi notare, mentre i sintomi cognitivi e psichiatrici in genere non sono abbastanza gravi da essere riconosciuti nelle prime fasi. L'esordio, la progressione e l'entità dei sintomi cognitivi e psichiatrici variano in modo significativo tra gli individui.
I più caratteristici sintomi fisici iniziali sono movimenti a scatti, casuali e incontrollabili chiamati "corea". La corea può essere inizialmente visibile come irrequietezza generale, piccoli movimenti involontari o incompleti, mancanza di coordinamento, rallentamento dei movimenti oculari saccadici. Queste anomalie motorie minori precedono in genere di almeno tre anni i più evidenti segni di disfunzione motoria. La chiara apparizione di sintomi come la rigidità, movimenti contorti o posture anomale appaiono quando la malattia progredisce. Questi sono segni che il sistema del cervello che regola il movimento è stato colpito. Le funzionalità psicomotorie diventano sempre più ridotte, in modo tale che qualsiasi azione che richiede il controllo muscolare viene influenzata. Le conseguenze più comuni sono instabilità, anormale espressione del viso e difficoltà nel masticare, deglutire e nel parlare. La difficoltà nel mangiare, comunemente può portare a perdita di peso e malnutrizione. Disturbi del sonno vengono considerati anch'essi dei sintomi. I pazienti pediatrici con malattia di Huntington presentano un decorso sintomatologico diverso, in quanto la malattia progredisce generalmente più velocemente e la corea si presenta fin dall'inizio con rigidità, essendo questo il sintomo dominante. Pure gli episodi convulsivi si presentano frequentemente.
| Irritabilità | 38–73% |
| Apatia | 34–76% |
| Ansia | 34–61% |
| Umore depresso | 33–69% |
| Disturbo ossessivo-compulsivo | 10–52% |
| Psicosi | 3–11% |
Le capacità cognitive vengono progressivamente compromesse. In particolare vengono colpite le funzioni esecutive che comprendono la pianificazione, la flessibilità cognitiva, il pensiero astratto, l'acquisizione delle regole, l'avvio di azioni appropriate e l'inibizione di azioni inappropriate. Col progredire della malattia tendono a comparire deficit di memoria. Vengono rilevati deficit di memoria a breve termine e difficoltà nella memoria a lungo termine, tra cui deficit nella memoria episodica (memoria della propria vita), nella memoria procedurale (memoria di come si esegue un'attività) e la memoria di lavoro. Le disabilità cognitive tendono a peggiorare nel tempo, fino a portare a uno stato di demenza. Quest'insieme di deficit è stato definito come "demenza sottocorticale" per distinguerle dagli effetti tipici delle demenze corticali, come ad esempio la malattia di Alzheimer.
Sono state riportate anche manifestazioni neuropsichiatriche, quali ansia, depressione, una riduzione delle emozioni (svuotamento affettivo), egocentrismo, aggressività e comportamenti compulsivi. Questi ultimi possono causare o peggiorare alcune dipendenze, tra cui alcolismo, ludopatia e l'ipersessualità. Sono state osservate anche difficoltà nel riconoscere le espressioni negative di altre persone. La prevalenza di questi sintomi è molto variabile negli studi effettuati, con tassi stimati tra il 33% e il 76%. Per molti malati e le loro famiglie, questi I sintomi sono tra gli aspetti più angoscianti della malattia, che spesso colpiscono la vita quotidiana e che costituiscono motivo di ricovero. I pensieri e i tentativi di suicidio sono più frequenti nei malati rispetto alla popolazione generale.
L'huntingtina mutante si esprime in tutto il corpo ed è correlata ad anomalie nei tessuti periferici che sono direttamente causate da tale espressione genica al di fuori del cervello. Queste anomalie comprendono: atrofia muscolare, insufficienza cardiaca, ridotta tolleranza al glucosio, perdita di peso, osteoporosi e atrofia testicolare.
Esami di laboratorio e strumentali
Strumenti di imaging biomedico, come la tomografia computerizzata (TC) e la risonanza magnetica (MRI), possono mostrare, all'inizio della malattia, atrofia del nucleo caudato. Tuttavia questo aspetto non è diagnostico per la condizione. Un'atrofia cerebrale generalizzata può essere vista nelle fasi avanzate della malattia. Tecniche di neuroimaging funzionale come la risonanza magnetica e la PET possono mostrare cambiamenti nell'attività cerebrale prima della comparsa dei sintomi fisici, ma sono strumenti sperimentali, e non ancora, al 2013, utilizzati in clinica.
Diagnosi differenziale
Circa il 99% delle diagnosi di malattia di Huntington che si basano sui sintomi tipici e su una storia familiare di malattia, vengono poi confermati da test genetici per evidenziare la ripetizione della sequenza trinucleotidica. La maggior parte dei casi restanti che non presentano alterazione genetica, vengono chiamati disturbi simil-Huntington. L'eziologia della maggior parte di queste condizioni rimane sconosciuta. Altre malattie autosomiche dominanti che possono essere erroneamente diagnosticate come malattia di Huntington sono atrofia dentato-rubra e neuroferritinopatia. Vi sono anche malattie autosomiche recessive che assomigliano alla malattia di Huntington. Esempi principali sono la corea acantocitosi, neurodegenerazione associata alla pantotenato chinasi e la sindrome di McLeod legata al sesso.
La corea può svilupparsi anche in seguito a patologie non genetiche, come ictus, lupus eritematoso sistemico (LES), sindromi paraneoplastiche, infezione da HIV, e come effetto collaterale di alcuni farmaci. Raramente nei bambini, in seguito a un'infezione da streptococco di gruppo A, può manifestarsi la corea di Sydenham, autolimitante.
Diagnosi precoce
La diagnosi medica dell'insorgenza della malattia di Huntington può essere fatta dopo la comparsa dei sintomi fisici specifici alla malattia. Il test genetico può essere usato per la conferma della diagnosi, se non vi è alcuna storia familiare della condizione. Anche prima della comparsa dei sintomi, i test genetici possono confermare se un individuo o un embrione possiede una copia con ripetizioni della sequenza trinucleotidica del gene HTT che causa la malattia. La diagnosi precoce ha forti implicazioni sulla psicologia di un individuo, sulla sua carriera lavorativa, sulle sue decisioni in merito al futuro, sul suo nucleo familiare e sulle sue relazioni. Nonostante la disponibilità di test pre-sintomatici, solo il 5% delle persone a rischio di ereditare la malattia decide di avvalersene.
Poiché la malattia di Huntington prevede un modello ereditario di tipo autosomico dominante, vi è una forte probabilità di sviluppare la condizione, nelle persone che presentano un rischio genetico. Il test genetico per la malattia consiste in un esame del sangue che conta il numero di ripetizioni CAG in ciascuno degli alleli HTT. Un risultato positivo non è considerato comunque una diagnosi, in quanto può essere ottenuto decenni prima che i sintomi abbiano inizio. Tuttavia, un test negativo significa che l'individuo non porta nel suo cariotipo la copia espansa del gene e quindi non svilupperà mai la malattia.
L'esecuzione di un test prima che i sintomi abbiano inizio, è un evento che può cambiare la vita e quindi una decisione molto personale. Il motivo principale per cui si consiglia di eseguire il test è per avere un aiuto nelle decisioni sulla carriera e sulla famiglia. Oltre il 95% delle persone a rischio di ereditare la malattia scelgono di non eseguire il test, soprattutto perché non esiste alcun trattamento. Una questione chiave è l'ansia che un individuo sperimenta per il non sapere se svilupperà la condizione, rispetto all'impatto di un risultato positivo. Indipendentemente dal risultato, i livelli di stress sono stati trovati più bassi due anni dopo l'esecuzione del test, ma il rischio di suicidio appare aumentato dopo un risultato positivo. Gli individui che si trovano nella condizione di non avere ereditato la malattia, possono sperimentare la colpa del sopravvissuto verso i membri della propria famiglia che ne sono invece colpiti. La consulenza genetica nella malattia è in grado di fornire informazioni, consigli e supporto sia per il processo decisionale sia per la fase successiva al risultato del test.
Counseling e le linee guida sull'uso dei test genetici per la malattia sono diventati modelli per le altre malattie genetiche, come le atassie cerebellari autosomiche dominanti. I test presintomatici per la malattia di Huntington hanno anche influenzato i test per altre malattie con varianti genetiche, come la malattia policistica renale autosomica dominante, la malattia di Alzheimer e il cancro alla mammella. La Genetics Quality Network European Molecular ha pubblicato uno schema annuale di valutazione esterna di qualità per i test genetici molecolari di questa malattia e questo ha permesso di mettere a punto le linee guida sulle migliori pratiche per i test genetici per la condizione e per l'assistenza al test e per la comunicazione dei risultati.
Diagnosi genetica preimpianto
Gli embrioni prodotti tramite fecondazione in vitro possono essere geneticamente testati per la malattia di Huntington utilizzando la diagnosi genetica preimpianto (PGD). Questa tecnica, in cui una o due cellule vengono estratte da un embrione e poi testate per l'anomalia genetica, può quindi essere utilizzata per garantire che embrioni presentanti geni con la malattia non vengono impiantati e pertanto la progenie non erediterà la malattia. Alcune forme di diagnosi genetica preimpianto consentono agli individui a rischio di avere figli sani senza rivelare il proprio genotipo e senza avere informazioni sul proprio rischio. Nei test di esclusione, il DNA degli embrioni viene confrontato con quello dei genitori e dei nonni per evitare l'ereditarietà della porzione cromosomica contenente il gene dei nonni colpiti dalla malattia. Nei test di non divulgazione, solo gli embrioni liberi da malattia vengono impiantati in utero, mentre il genotipo dei genitori e, quindi, il loro rischio di sviluppare la condizione non viene mai reso noto.
Diagnosi prenatale
È possibile anche ottenere una diagnosi prenatale su di un embrione o su un feto nel grembo materno, utilizzando materiale genetico fetale acquisito attraverso il prelievo di villi coriali (villocentesi) o di liquido amniotico (amniocentesi). Anche questa tecnica può essere accoppiata con i test di esclusione per evitare la divulgazione del genotipo dei genitori. La diagnosi prenatale viene eseguita con l'intenzione che se il feto viene trovato con le ripetizioni nel gene HTT (o, nei test di esclusione, si trova che è ad 'alto rischio'), la gravidanza verrà terminata.
Trattamento
Non esiste una cura per la malattia di Huntington, ma vi sono trattamenti disponibili per ridurre la gravità di alcuni dei suoi sintomi. Per molti di essi, non vi sono studi clinici completi per confermarne la loro efficacia. Come la malattia progredisce, declina sempre di più la capacità di prendersi cura di sé stessi e diviene necessaria un'assistenza multidisciplinare. Anche se vi sono stati relativamente pochi studi sugli esercizi fisici e sulle terapie che possano aiutare a riprendere i deficit cognitivi, vi sono alcune prove a favore dell'utilità della terapia fisica, della terapia occupazionale e della logopedia.
La perdita di peso e la difficoltà di mangiare per via della disfagia dovuta alla mancanza di coordinazione muscolare, sono caratteristiche comuni nella malattia, e rendono pertanto la gestione della nutrizione un aspetto sempre più importante con l'avanzare della malattia. Agenti addensanti possono essere aggiunti ai liquidi per renderli più densi e quindi più sicuri da deglutire. Può essere d'aiuto anche ricordando al paziente di mangiare lentamente e di prendere piccoli pezzi di cibo in bocca per evitare il soffocamento. Se mangiare diventa troppo pericoloso o scomodo, si può ricorrere a una gastrostomia endoscopica percutanea. Si tratta di un tubo per l'alimentazione, fissato in modo permanente attraverso l'addome e verso lo stomaco, che così riduce il rischio di aspirazione del cibo e fornisce una migliore gestione della nutrizione. La valutazione e la gestione da parte di logopedisti con esperienza nella malattia di Huntington, è fortemente raccomandata.
I pazienti con malattia di Huntington possono ricorrere a un fisioterapista per trattare in modo non invasivo e senza farmaci i sintomi fisici. I fisioterapisti possono valutare il rischio di caduta e le strategie più idonee per prevenirle, nonché possono provvedere a un rafforzamento muscolare, allo stretching e a esercizi cardiovascolari. Ausili per la deambulazione possono essere anche prescritti a seconda dei casi. I fisioterapisti prescrivono anche esercizi di respirazione e tecniche di liberazione delle vie aeree quando si va incontro allo sviluppo di problemi respiratori. Le linee guida terapeutiche per la fisioterapia nella malattia di Huntington sono state formulate dalla European Huntington's Disease Network. L'obiettivo degli interventi riabilitativi precoci è la prevenzione della perdita delle funzioni. La partecipazione a programmi di riabilitazione durante la prima fase della malattia può essere utile in quanto si traduce in un mantenimento più a lungo termine delle capacità motorie e delle prestazioni funzionali. La riabilitazione durante la fase tardiva mira invece a compensare le perdite motorie e funzionali. Per una gestione indipendente a lungo termine, il terapeuta può proporre programmi di esercizi personalizzati da effettuarsi a domicilio.
Trattamento farmacologico
Sono stati segnalati numerosi farmaci in grado di produrre benefici in animali, tra cui la creatina, il coenzima Q10 e la minociclina antibiotico. Alcuni di questi sono stati poi testati su esseri umani in vari studi clinici, ma finora nessuno si è dimostrato efficace. Nel 2010, la minociclina è risultata inefficace per gli esseri umani. Grandi studi osservazionali, che coinvolgono volontari umani, hanno rivelato tuttavia molte informazioni sulla fisiopatologia della malattia di Huntington fondamentali per futuri trial clinici.
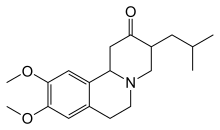
La tetrabenazina è stata approvata nel 2008, negli Stati Uniti d'America, per il trattamento della corea nella malattia di Huntington. Altri farmaci che aiutano a ridurre la corea comprendono le famiglie dei neurolettici e delle benzodiazepine. Composti come amantadina o remacemide sono, al 2013, ancora in fase di studio, ma hanno mostrato risultati preliminari positivi. L'ipocinesia e la rigidità, soprattutto nei casi giovanili, possono essere trattate con farmaci antiparkinsoniani, mentre le ipercinesiaemiocloniche possono essere trattate con l'acido valproico.
I sintomi psichiatrici possono essere trattati con farmaci simili a quelli utilizzati per la popolazione generale. Gli inibitori selettivi della ricaptazione della serotonina e la mirtazapina sono stati raccomandati per la depressione, mentre i farmaci antipsicotici atipici sono raccomandati per la cura della psicosi e dei problemi comportamentali. Il consulto specialistico neuropsichiatrico, viene raccomandato per i pazienti che necessitano di un trattamento a lungo termine con più farmaci in combinazione.
Stato della ricerca

La ricerca sulla fisiopatologia della malattia di Huntington si è concentrata sull'identificazione del funzionamento della HTT, come la mHtt differisce o come interferisce con essa e come la malattia interferisca sul funzionamento del cervello. La ricerca è stata condotta con metodi in vitro, modelli animali e volontari umani. I modelli animali sono fondamentali per la comprensione dei meccanismi fondamentali che causano la malattia e per sostenere le prime fasi di sviluppo di un farmaco. Animali a cui è stata indotta chimicamente la lesione nel cervello, mostrano sintomi simili alla malattia, ma tuttavia non ne hanno imitato le caratteristiche progressive. L'identificazione del gene della malattia ha permesso lo sviluppo di numerosi modelli di animali transgenici tra cui vermi nematodi, Drosophila melanogaster, topi, ratti, pecore, maiali e scimmie che esprimono la huntingtina mutante e sviluppano una progressiva neurodegenerazione e sintomi simili alla condizione.
Tre approcci generali sono sotto studio per tentare di rallentare la progressione della malattia di Huntington: ridurre la produzione della proteina mutante, migliorare la capacità delle cellule di sopravvivere ai suoi diversi effetti nocivi e sostituire i neuroni persi.
- Ridurre la produzione di huntingtina
Poiché la malattia è causata da un singolo gene dominante codificante una proteina tossica, il silenziamento genico mira a ridurre la produzione della proteina mutante. Esperimenti effettuati su topi hanno dimostrato che quando l'espressione della mHTT è ridotta, i sintomi migliorano. La sicurezza del silenziamento genico deve essere ora, al 2013, dimostrata nei cervelli di notevoli dimensioni come negli umani e nei primati.
- Migliorare la sopravvivenza cellulare
Tra gli approcci volti a migliorare la sopravvivenza delle cellule in presenza della huntingtina mutante, vi è la correzione della regolazione trascrizionale utilizzando inibitori dell'istone deacetilasi, l'aggregazione modulare di huntingtina, l'aumento del metabolismo e della funzione mitocondriale e il ripristino delle disfunzioni sinaptiche.
- Sostituzione neuronale
La terapia con cellule staminali consiste nel sostituire i neuroni danneggiati, grazie al trapianto di cellule staminali, nelle regioni colpite del cervello. Gli esperimenti hanno dato risultati contrastanti con questa tecnica nei modelli animali e negli studi clinici preliminari. Qualunque sia il loro futuro potenziale terapeutico, le cellule staminali sono già uno strumento prezioso per lo studio in laboratorio della malattia.
Prognosi
Il numero delle ripetizioni trinucleotidiche incide per il 60% sulla sintomatologia, sull'età di esordio e sulla progressione della malattia. Un numero elevato di ripetizioni comporterà una più precoce età di insorgenza della malattia e una progressione più rapida dei sintomi. Gli individui con più di sessanta ripetizioni spesso sviluppano la malattia prima dei 20 anni, mentre quelli con meno di 40 ripetizioni potrebbero non mostrare alcun sintomo evidente. Gli aspetti che incidono ulteriormente sulla malattia sono i fattori ambientali e gli altri geni che influenzano l'andamento della malattia.
La speranza di vita in un malato è in genere di circa 20 anni dopo che si sono resi visibili i primi sintomi.
Bibliografia
- Karen Bellenir (a cura di), Huntington Disease, in Genetic Disorders Sourcebook, 3rd, Detroit, Omnigraphics, 2004, pp. 159–179, ISBN 0-7808-0742-1.
- Harper P, Huntington's disease: a historical background, in Bates G, Harper P, and Jones L (a cura di), Huntington's Disease – Third Edition, Oxford, Oxford University Press, 2002, pp. 3–24, ISBN 0-19-851060-8.
- Harper P, Huntington's disease: a historical background, in Bates G, Harper P, and Jones L (a cura di), Huntington's Disease – Third Edition, Oxford, Oxford University Press, 2002, pp. 3–24, ISBN 0-19-851060-8.
- Wexler A, Wexler N, The Woman Who Walked Into the Sea. Huntington's and the Making of a Genetic Disease, Yale University Press, 2008, pp. 288, ISBN 978-0-300-10502-5.
- Driver-Dunckley E, Caviness JN., Huntington's disease, in Schapira AHV (a cura di), Neurology and Clinical Neuroscience, Mosby Elsevier, 2007, pp. 879–885, ISBN 978-0-323-03354-1.
- E Passarge, Color Atlas of Genetics, 2nd, Thieme, 2001, pp. 142, ISBN 0-86577-958-9.
- Purves D, Augustine GA, Fitzpatrick D, Hall W, LaMantia A-S, McNamara JO, Williams SM, Modulation of Movement by the Basal Ganglia – Circuits within the Basal Ganglia System, in Purves D (a cura di), Neuroscience, 2nd, Sunderland, MA, Sinauer Associates, 2001, ISBN 0-87893-742-0. URL consultato il 1º aprile 2009.
- Kremer B, Clinical neurology of Huntington's disease, in Bates G, Harper P, and Jones L (a cura di), Huntington's Disease – Third Edition, Oxford, Oxford University Press, 2002, pp. 28–53, ISBN 0-19-851060-8.
- Crauford D and Snowden J, Neuropyschological and neuropsychiatric aspects of Huntington's disease, in Bates G, Harper P, and Jones L (a cura di), Huntington's Disease – Third Edition, Oxford, Oxford University Press, 2002, pp. 62–87, ISBN 0-19-851060-8.
- Harper P, The epidemiology of Huntington's disease, in Bates G, Harper P, and Jones L (a cura di), Huntington's Disease – Third Edition, Oxford, Oxford University Press, 2002, pp. 159–189, ISBN 0-19-851060-8.
Altri progetti
-
 Wikiquote contiene citazioni sulla malattia di Huntington
Wikiquote contiene citazioni sulla malattia di Huntington
-
 Wikimedia Commons contiene immagini o altri file su malattia di Huntington
Wikimedia Commons contiene immagini o altri file su malattia di Huntington
Collegamenti esterni
- (EN) Malattia di Huntington, su Enciclopedia Britannica, Encyclopædia Britannica, Inc.
- Huntington Onlus, la rete italiana della malattia di Huntington, su http://www.huntington-onlus.it/
- Associazione italiana della Corea di Huntington Aich Milano Onlus, su aichmilano.it.
- Associazione italiana della Corea di Huntington Aich Roma Onlus, su aichroma.com.
- Associazione Italiana della Corea di Huntington Aich Napoli Onlus, su aichnapoli.it.
- HDBUZZ - Novità dalla ricerca mondiale sulla Malattia di Huntington, su it.hdbuzz.net.
- HDYO - Organizzazione internazionale per i giovani che hanno a che fare con l'Huntington, su it.hdyo.org.
- Lega italiana per la ricerca sulla malattia di Huntington - Fondazione privata
- (EN) * (EN) Huntington Project, su huntingtonproject.org. URL consultato il 23 febbraio 2009 (archiviato dall'url originale il 24 ottobre 2008).
- Huntington Days, le giornate di consapevolezza sulla malattia di Huntington, su http://www.huntington-onlus.it/cosa-facciamo/huntingtons-days/
| Controllo di autorità | Thesaurus BNCF 55969 · LCCN (EN) sh85063158 · GND (DE) 4026223-6 · BNE (ES) XX548572 (data) · BNF (FR) cb12257151p (data) · J9U (EN, HE) 987007533753105171 |
|---|